



どのような病気?
1p36欠失症候群は、成長障害、精神発達遅滞、てんかん、特徴的な顔立ちなどを主な症状とする遺伝性疾患です。
この病気では、赤ちゃんの頃から、筋緊張低下や哺乳不良などの症状が現れます。精神運動発達遅滞はほとんどの患者さんで見られます。発達の遅れによる知的障害は軽度から重度までさまざまです。発話障害や言語発達の遅延によって、ほとんど話せるようにならない場合もあります。一方で、遅れながらも少しずつ発達が伸びていき、構音障害を示しながらも会話が可能になる場合もあるとされます。
この病気を持つ人の顔立ちの特徴は、まっすぐな眉、落ちくぼんだ目、とがったあご、顔の中央がくぼんだように見える(顔面中部後退)などです。
その他の症状として、難治性てんかん、先天性心疾患を合併する場合もあります。斜視、白内障、難聴など、視覚や聴覚に問題が起こることもあります。また、骨格、消化器系、腎臓、性器に異常が起こる場合もあります。
| 1p36欠失症候群で見られる症状 |
|---|
高頻度に見られる症状 まっすぐな眉、くぼんだ目、顔面中部後退、広い鼻梁、長い人中(唇上部の溝)、とがったあご、短指症、指の屈指症(指をまっすぐに伸ばせない)、短い足、発育不良、歩行障害、筋緊張低下、脳室拡大、脳梁欠損症、大脳皮質萎縮、全体的な発達遅延、知的障害、神経性言語障害、発話と言語発達の遅延、発語欠損 |
良く見られる症状 短頭症、小頭症、眉毛形態異常、内眼角贅皮(ないがんかくぜいひ、上まぶたが目頭部分を覆う)、低い鼻梁、平らな鼻堤、口の幅が狭い、低い位置で後方に回転した耳、小指の湾曲、心血管系の形態異常、便秘、胃食道逆流、嚥下障害、幼少期の摂食困難、頭蓋骨縫合閉鎖遅延、てんかん、常同行動、自傷行為、視力の異常、重度の遠視、斜視、自閉症、行動異常 |
しばしば見られる症状 前額部の突出、小耳症、首の形態異常、皮膚の異常な水ぶくれ、しみ(斑)、下肢の非対称性、足の多指症、女性外性器異常、停留精巣、陰茎の形成不全、尿道下裂、肛門の異常、全身性多毛症、低身長、11対の肋骨(肋骨が少ない)、二分肋骨、肋骨の融合、脊柱側弯症、脊柱管狭窄症、脊柱後湾症、股関節形成不全、関節のこわばり、拡張型心筋症、心室中隔形態の異常、心臓弁膜形態の異常、毛細血管拡張症、弓部大動脈瘤、ファロー四徴症、動脈管開存症、幽門狭窄、腸の形態異常、腎臓の異常、肝臓の異常、脾臓の異常、輪状膵(膵臓の一部が十二指腸に輪状に取り巻き十二指腸を閉塞する)、腎嚢胞、脂肪肝、水腎症、性腺機能低下症、甲状腺機能低下症、免疫系の異常、脳神経麻痺、片麻痺、ミオパチー、神経芽細胞腫、過食、肥満、眼球白子症、眼球振とう、視神経萎縮、伝音性難聴、感音性難聴、白内障、視力障害 |
1p36欠失症候群は、世界では5,000人から1万人に1人の頻度であると推定されていますが、この病気であると気付かれず、診断を受けていない人がいるのではないかと考えられています。難病情報センターによれば、日本では調査の結果、およそ2万5,000人から4万人に1人の頻度と考えられ、そこから年間10~20人程度の患者さんの出生があると推定されています。
この病気は、民族や人種による発症頻度には差がありませんが、男性よりも女性の方が多いことがわかっており、男性3割、女性7割の比率で認められます。
1p36欠失症候群は指定難病対象疾病(指定難病197)となっています。
何の遺伝子が原因となるの?
1p36欠失症候群は1番染色体の短腕の末端、1p36領域に欠失(一部の遺伝子とともになくなること)があり、欠失した部分に存在していた遺伝子の機能が失われることでこの病気が引き起こされます。欠失の大きさは患者さんによって異なり、失われた遺伝子の数や種類も異なることがそれぞれの症状の違いや重症度に関わっていると考えられます。しかし、この病気のそれぞれの症状と関連することが確定的である遺伝子や、この病気を引き起こす詳細なメカニズムはまだわかっていません。
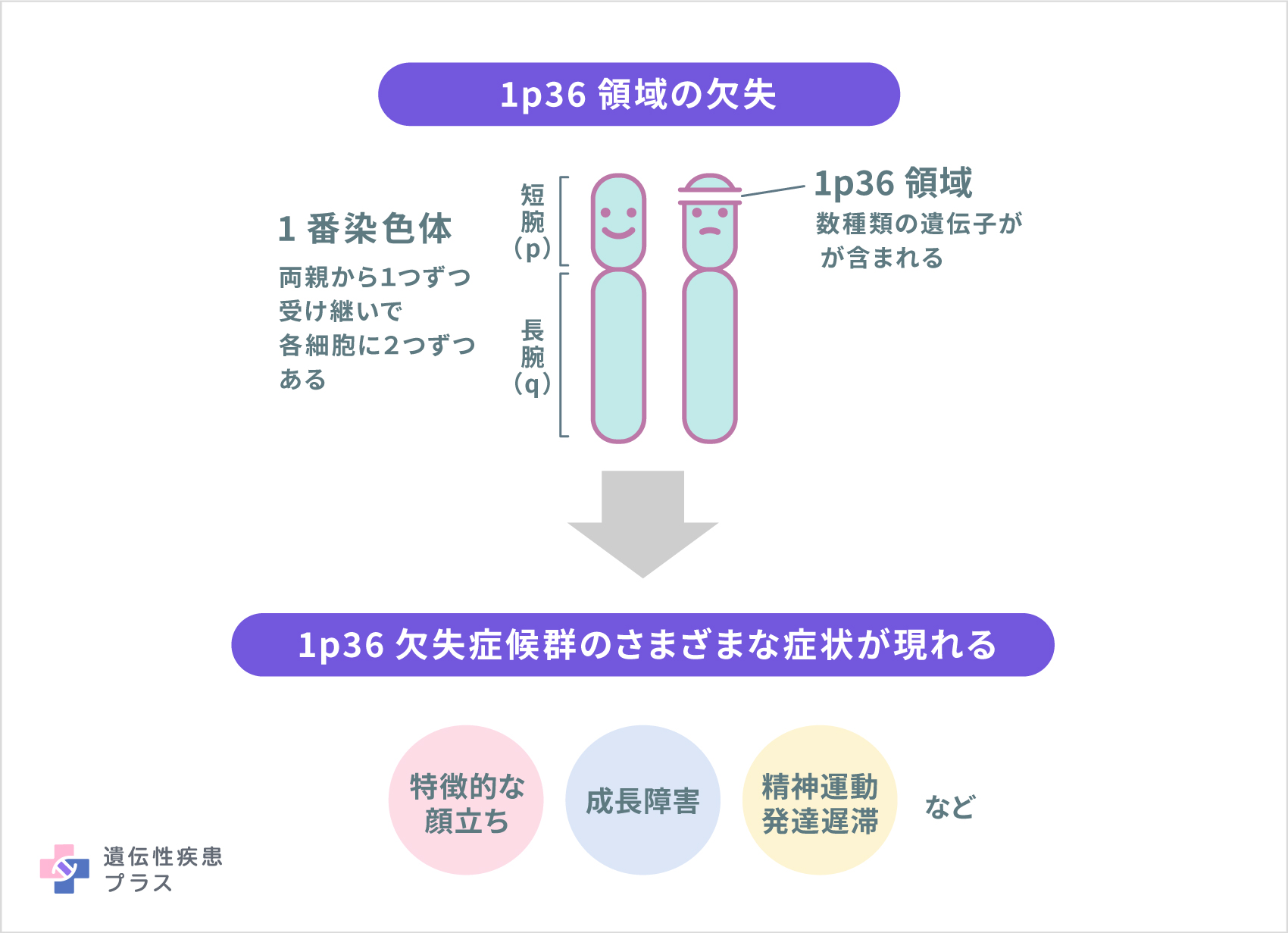
1p36欠失症候群において、これまでに報告されたほとんどの患者さんでは、両親がこの病気を発症していません。半数以上の患者さんは、生殖細胞(卵子または精子)が形成される時や胎児期の発育初期にランダムに起こる染色体欠失によって発症した孤発例です。
一方で、この病気の一部の人は、親が均衡転座と呼ばれる特別な染色体の状態を保持しており、それを受け継ぐことにより発症していることがあります。転座とは、ヒトが持つ2本の染色体の一部が切断され、断片が入れ替わって他の染色体に結合することをいいます。しかし染色体の一部が入れ替わっても、すべての遺伝子が揃っている場合、その人には病気の症状が現れないことがあり、これを均衡型転座と呼びます。均衡型転座をもつ親が子どもを持つ場合に染色体の分布が変わり遺伝情報の過不足が生じることがあります(不均衡型転座)。不足となった領域が1p36領域だった場合に、この病気が引き起こされます。
どのように診断されるの?
1p36欠失症候群の診断は、臨床所見と遺伝学的検査から行われます。
重要な症状として、1)精神発達遅滞(IQ70未満)、2)特徴的な顔立ち(まっすぐな眉毛、落ちくぼんだ眼、眼間狭小、とがった顎)の2つが見られ、遺伝学的検査によって染色体1p36領域の欠失が認められた場合に、この病気の診断が確定となります。
どのような治療が行われるの?
1p36欠失症候群における根本的な治療法はまだ見つかっておらず、それぞれの症状に応じた対症療法がとられます。発達の遅れや、筋緊張低下に関しては、乳幼児期からの療育訓練が行われます。てんかん発作には、てんかん薬が処方されることがあります。
また、患者さんの家族に対して、遺伝学的診断に基づく遺伝カウンセリングが勧められることもあります。
どこで検査や治療が受けられるの?
日本で1p36欠失症候群の診療を行っていることを公開している、主な施設は以下です。
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
難病の患者さん・ご家族、支えるさまざまな立場の方々とのネットワークづくりを行っている団体は、以下です。





