



どのような病気?
5p欠失症候群は、5番染色体の一部の欠失を原因とする遺伝性疾患で、主な症状として、全体的な成長や精神運動発達の遅れ、特徴的な顔立ちのほか、赤ちゃんの頃の甲高い声の泣き声などがみられます。
甲高い泣き声は、新生児期から乳児期において高い頻度で認められます。この特徴から、海外では「Cat cry syndrome」「Cri-du-chat syndrome」という別名で呼ばれることもあります。日本では、この別名(猫鳴き症候群)を、推奨しない声が挙がっています。その他にも、2,500g未満の低出生体重や成長障害、筋緊張低下も赤ちゃんの時に見られる症状です。心臓に異常を持って生まれてくる場合もあります。
顔立ちや見た目の特徴として、頭のサイズが小さい(小頭症)、丸い顔、大きく開いた目、眼と眼の間が広い(眼間開離)、顎が小さい、目頭部分を上まぶたが覆う(内眼角贅皮・ないがんかくぜいひ)、低い位置にある耳(耳介低位)などがあります。思春期から成人期以降は、小頭がはっきりするようになり面長の顔や大きな口が目立つなど特徴は成長とともに変化します。筋緊張は逆に亢進するようになります。
多くの場合、この病気では乳幼児期から、知的障害を含む精神運動発達遅滞が見られます。具体的には、頭を動かす、座る、歩くなど、精神的かつ筋肉的な活動を必要とする能力の獲得の遅れが見られます。これまでの報告では、この病気の半数の子どもが5歳までには自分で着替えることができるようになっています。知的障害は、中程度から重度と言われており、特に言葉の発達が遅れます。どちらかというと意思疎通よりも言葉を理解することの方が得意であるとされます。多動や自傷行為などの行動の問題が見られる場合もあります。
また、心臓の他にも循環器、泌尿生殖器、筋骨格、眼科、耳鼻科などの多領域に渡る合併症がみられることもあり、手術が必要となる場合もあります。
5p欠失症候群で見られる症状 |
|---|
高頻度に見られる症状 小頭症、丸い顔、内眼角贅皮、幅の広い鼻梁(びりょう)、低い位置で後方に回転した耳、小さく後退している下顎、声の異常、猫の鳴き声、甲高い声、筋緊張低下、重度の知的障害、重度の精神運動発達遅滞 |
よく見られる症状 眼間開離、眼瞼裂斜下(目尻が標準よりも下がっている)、高口蓋、短い首、小さな手、低身長、脊柱側弯症、(胎児期の)子宮内発育遅延 |
しばしば見られる症状 耳介前肉柱、合指症(隣り合う指がくっついている状態)、鼠径ヘルニア、関節過剰屈曲、反復性骨折、骨密度の異常、心血管系形態異常 |
5p欠失症候群は、世界で出生1万5,000~2万人に1人の頻度で発症すると推定されています。民族や人種による発症頻度の差は確認されていません。また、この病気は男性よりもわずかに女性に多くみられるという報告があります。
何の遺伝子が原因となるの?
5p欠失症候群は、5番染色体の短腕(p)の末端領域の一部の欠失によって引き起こされます。欠失とは一部の遺伝子とともにその領域がなくなっていることで、欠失部分の大きさはそれぞれの患者さんによって異なります。欠失した部分が大きいほど知的障害や発達の遅れが重症となるという報告もあります。
この病気の症状や特徴は、5番染色体上の欠失部分にある複数の遺伝子の機能と関連していると考えられます。この領域におけるそれぞれの遺伝子の欠失がどのようにこの病気の症状と関連しているかは、まだ研究が進められており詳細ははっきりしていません。
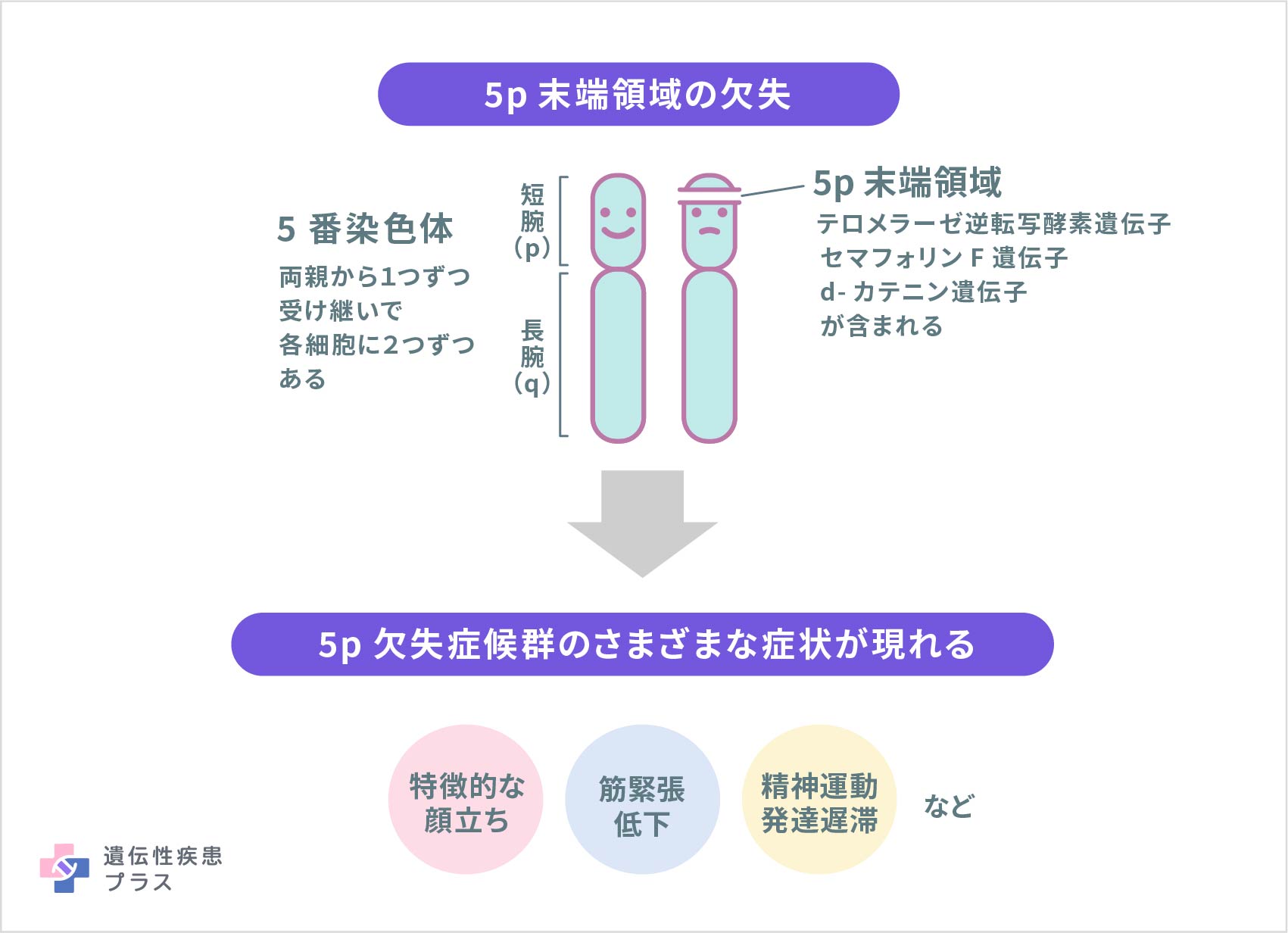
米国希少疾病協議会(NORD)によると、これまでの研究で5p13.33領域にあるテロメラーゼ逆転写酵素遺伝子や5p15.2領域にあるセマフォリンF遺伝子の欠失と、この病気の顔立ちや見た目のさまざまな特徴との関連が示唆されています。また、5p15.2領域にあるd-カテニン遺伝子は神経細胞の発達初期に働くタンパク質の設計図であり、その欠失と重度の知的障害との関連が示唆されています。
5p欠失症候群では、約8割で両親はこの病気を発症しておらず孤発例として発症します。このような染色体欠失は、生殖細胞(卵子または精子)の形成時や胎児期の発育初期にランダムに起こります。このほか、約1割において、均衡型転座とよばれる特別な染色体の状態(異なる染色体で一部が入れ替わっているが全体で見ると重複や欠失が無い状態)により病気を発症していない親から、変化した染色体を引き継いだ子どもが、不均衡型転座(一部重複や欠失がある状態)となることでこの病気を発症することがあります。
どのように診断されるの?
5p欠失症候群は、1)新生児期から乳児期の甲高い啼泣(ていきゅう・声をあげて泣くこと)、2)小頭症、3)成長障害の3つの臨床所見を乳幼児期に全て認め、遺伝学的検査によって5番染色体の5p15領域の欠失が認められた場合に診断されます。
どのような治療が行われるの?
この病気の根本的な治療法は、まだ見つかっていません。そのため、それぞれの症状に応じた対症療法が行われます。心疾患やその他の合併症に対しては手術や薬物療法の治療が行われるほか、新生児期には呼吸障害や哺乳障害の治療、成長障害の管理が行われます。年代ごとに注意すべき問題点が変わってくるため、それぞれに応じた対応や治療が行われます。
どこで検査や治療が受けられるの?
日本で5p欠失症候群の診療を行っていることを公開している、主な施設は以下です。
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
5p欠失症候群の患者会で、ホームページを公開しているところは、以下です。





