



どのような病気?
アラン・ハーンドン・ダドリー症候群は脳の発達障害によって知的障害や運動障害を生じる遺伝性疾患で、ほとんどは男児に発症します。アラン・ハーンドン・ダドリー症候群の患者さんの多くには、筋緊張低下や筋肉の発達低下が見られます。また、関節の変形による可動域の制限、筋肉の痙縮(けいしゅく、筋肉の緊張が亢進した状態)、筋力低下、手足の不随意運動(自分の意思に基づかない運動)などによって運動機能が障害され、助けを借りずに自力で歩くことが困難となります。
アラン・ハーンドン・ダドリー症候群では、神経学的な異常や甲状腺機能の異常によるさまざまな症状が見られます。胎児期においては、胎動低下(胎児が動く頻度が低下)や早産、羊水過多など、新生児期には低体重、低身長、小頭症、知的障害などが見られます。神経学的な異常による症状としては、筋緊張低下、ジストニア(意思にかかわらず体がこわばる症状)、てんかん発作などがあります。
アラン・ハーンドン・ダドリー症候群で見られる症状 |
|---|
高頻度に見られる症状 体幹部筋緊張低下、知的障害 |
よく見られる症状 錐体路(すいたいろ)の機能障害による神経学的異常(錐体路は大脳皮質から脊髄につながる運動神経伝導路)、甲状腺機能異常、運動失調、脳萎縮、不随意運動、BMI低下(やせ)、歩行能力遅滞、髄鞘形成遅延(神経の情報伝達を早くする髄鞘の形成が遅延すること)、会話および言語発達遅延、ジストニア、乳児期の発育不全、乳児期の摂食障害、反射亢進、後湾症(脊柱が後ろに湾曲した状態)、長い顔、筋障害性顔貌(こけた頬、眼瞼下垂、無表情などの特有の顔立ち)、ほっそりした顔、漏斗胸(ろうときょう、中央部に凹みがある胸)、偏平足、外反足、骨格筋委縮、痙縮 |
しばしば見られる症状 錐体外路性運動機能障害(錐体外路は運動に関わる神経経路)、足クローヌス(足の律動的な異常運動)、バビンスキー兆候(足の裏を強くこすると足の親指が反る脊髄反射)、腱反射亢進、停留睾丸(睾丸が陰嚢の中に降りていない状態)、胎動減少、関節拘縮、全身筋力低下、四肢筋緊張亢進、小頭症、新生児筋緊張低下、眼振(意思とは関係なく眼球が動くこと)、頭部の姿勢維持困難、新生児黄疸持続、てんかん発作、低身長、けいれん性四肢麻痺 |
まれに見られる症状 ジスキネジア(自分では止められない不規則な体の動き)、大頭症、羊水過多、早産、妊娠週数に対して小さい胎児 |
アラン・ハーンドン・ダドリー症候群の発症頻度は不明ですが、米国の遺伝性疾患情報サイト「GeneReviews」によると、約15年間の調査により世界で160人以上の患者さんが確認されています。
アラン・ハーンドン・ダドリー症候群(MCT8欠損症)は、この疾患を含む11種類の疾患の総称である「先天性大脳白質形成不全症」として、指定難病対象疾病(指定難病139)および小児慢性特定疾病の対象疾患となっています。
何の遺伝子が原因となるの?
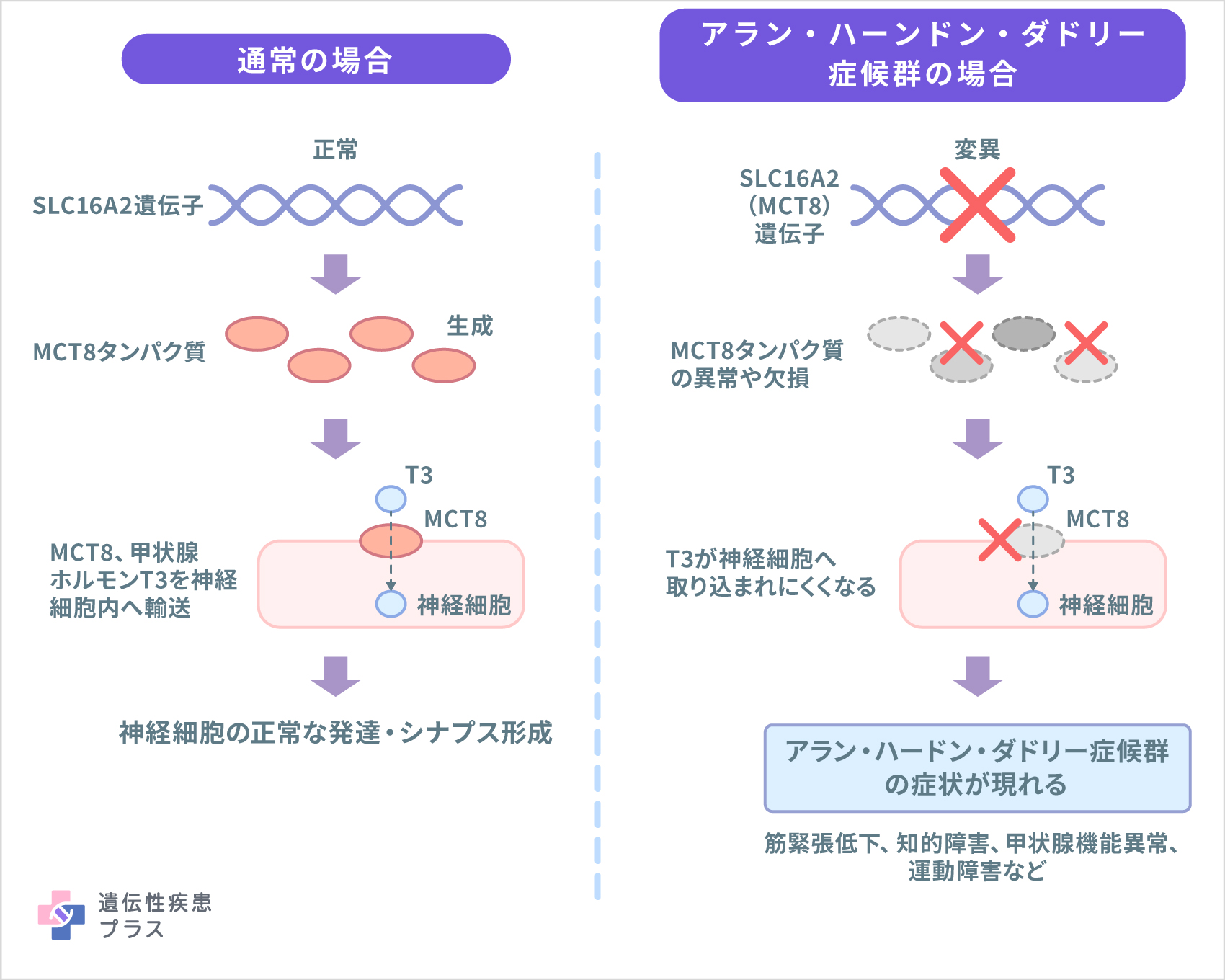
アラン・ハーンドン・ダドリー症候群の原因はSLC16A2という遺伝子の変異によります。この遺伝子はMCT8とも呼ばれます。神経細胞の発達や神経細胞間の接合部であるシナプスの形成においては、トリヨードチロニン(T3)という甲状腺ホルモンを神経細胞内に取り込む必要があります。SLC16A2によって産生されるタンパク質はT3を神経細胞内に輸送する作用を持ち、神経細胞の正常な発達やシナプスの形成に重要な役割を担っています。SLC16A2遺伝子の変異によって、T3の神経細胞への取り込みが低下することで神経細胞やシナプスの発達が障害され、アラン・ハーンドン・ダドリー症候群が発症すると考えられています。
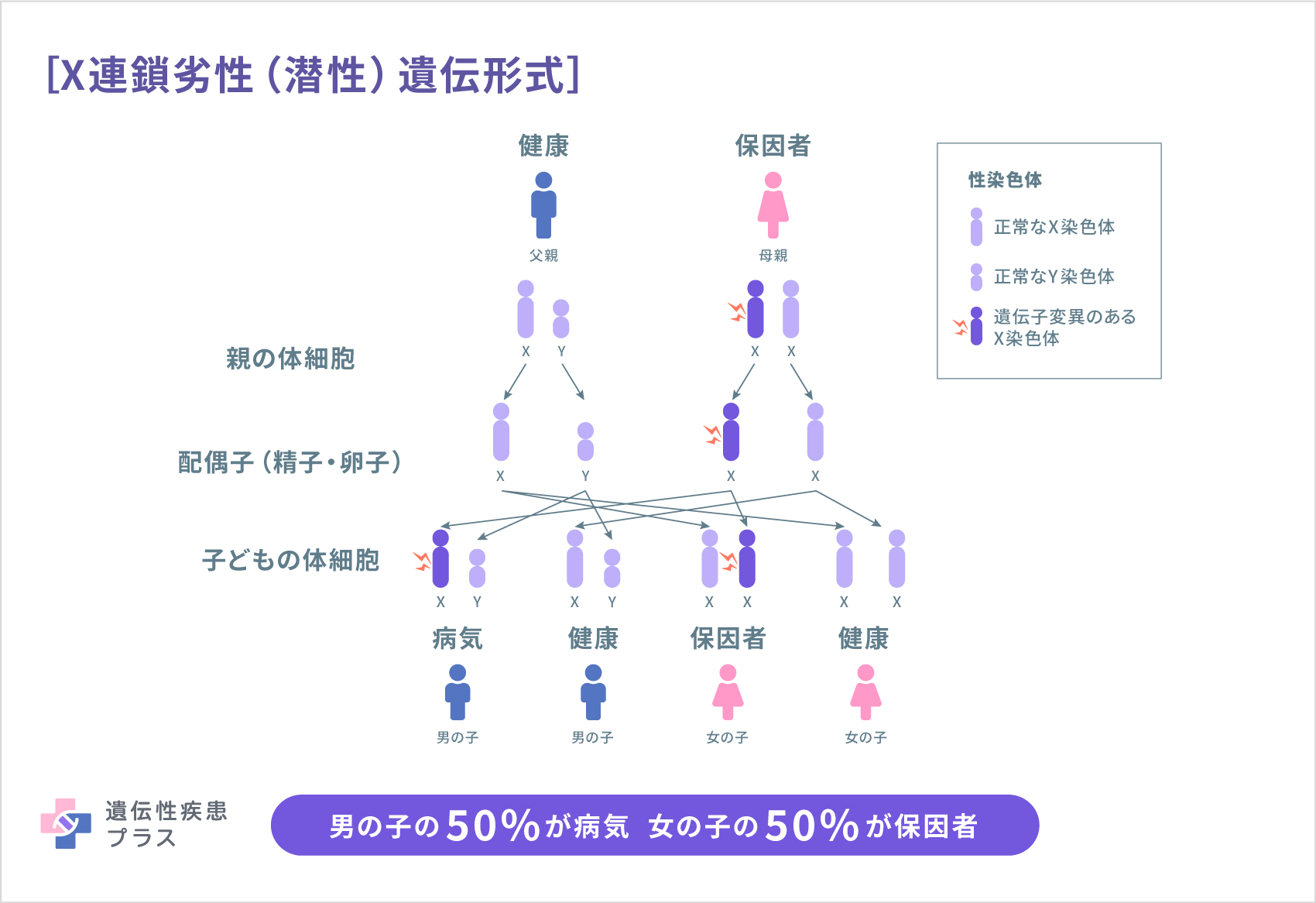
アラン・ハーンドン・ダドリー症候群は、X連鎖劣性(潜性)遺伝形式で遺伝します。アラン・ハーンドン・ダドリー症候群の原因であるSLC16A2遺伝子はX染色体にあります。ヒトの性染色体にはX染色体とY染色体の2種類があり、男性はXY、女性はXXの組み合わせです。男性では1本しかないX染色体のSLC16A2遺伝子に変異があるとアラン・ハーンドン・ダドリー症候群を発症しますが、女性は2本あるX染色体のうち片方のSLC16A2遺伝子に異常があっても、もう1本が機能を補完するため発症しません(この遺伝子異常があっても発病しない状態を保因者と言います)。男性のX染色体は母親から受け継ぎ、女性のX染色体は両親から1つずつ受け継ぎます。このため、父親がアラン・ハーンドン・ダドリー症候群の場合、子どもが男性の場合は発病せず、女性の場合は必ず保因者になります。また、保因者の母親から生まれた男児は2分の1の確率で発症し、女児は2分の1の確率で保因者になります。
どのように診断されるの?
難病情報センターでは、アラン・ハーンドン・ダドリー症候群について以下の診断基準が示されています。
下記Iのうち①を含む2つ以上とIIの①および②または③を満たすものをアラン・ハーンドン・ダドリー症候群と診断する。
I 主要臨床症状
①痙性四肢麻痺あるいは下肢麻痺
②眼振
③精神運動発達遅滞
④小脳障害:体幹・四肢の失調症状、企図振戦(目標物に手が届きそうになると強くなる体の震え)、小児期には測定障害(手足の運動を目的のところで止めることができない障害)、変換障害(身体的な症状が心理的な要因によって引き起こされる精神疾患)、緩弱言語(思考がまとまりを欠いて、言語表現が一貫性や論理性に乏しい状態)など
⑤基底核障害:固縮、ジストニア
II 重要な検査所見
①MRI画像所見:T2強調画像で白質にびまん性の高信号領域(脱髄性疾患の所見のあるものは除外する)
②甲状腺ホルモン検査にて、T4低値、T3高値、TSHは正常値上限
③遺伝子解析:SLC16A2異常
どのような治療が行われるの?
どこで検査や治療が受けられるの?
日本でアラン・ハーンドン・ダドリー症候群の診療を行っていることを公開している、主な施設は以下です。
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
難病の患者さん・ご家族、支えるさまざまな立場の方々とのネットワークづくりを行っている団体は、以下です。
参考サイト
- 難病情報センター
- 小児慢性特定疾病情報センター 先天性大脳白質形成不全症
- MedlinePlus
- Genetic and Rare Diseases Information Center
- GeneReviews






