



どのような病気?
アルポート症候群は、慢性腎炎、難聴、目の異常を特徴とする遺伝性疾患です。
アルポート症候群は多くの場合、幼少期までに血尿が出始めます。この血尿は、慢性腎炎によるものですが、通常見た目にはわからない程度であるため、尿検査で指摘されます。ただし風邪をひいたりした際に、赤っぽい尿が出て気付く場合もあります。この腎障害は年齢とともにゆっくりと進行していきます。徐々にタンパク尿が出るようになり、ネフローゼ症候群を起こす場合もあります。さらに進行すると腎臓が機能しなくなり、末期腎不全となります。アルポート症候群の症状には男女差があり、女性より男性で重症の人が多いことがわかっています。最も頻度の高い「COL4A5遺伝子の異常に伴うX染色体連鎖型アルポート症候群」では40歳までに男性の9割程度、女性の1割程度が、末期腎不全となります。男性の末期腎不全到達平均年齢は25歳くらいと報告されています。一方、女性では軽度の血尿やタンパク尿のみが見られる程度にとどまる人も多くいます。
難聴は、幼少期までに認められることはありませんが、「COL4A5遺伝子の異常に伴うX染色体連鎖型アルポート症候群」では、多くの男性で10歳を過ぎた頃から内耳の異常によって引き起こされる感音性難聴(両側性、進行性)が認められるようになります。男性は最終的に8割程度の人で難聴が認められます。女性は2割程度で認められます。
目の異常としては、皮質部白内障、円錐水晶体、水晶体脱臼などが認められる場合があります。「COL4A5遺伝子の異常に伴うX染色体連鎖型アルポート症候群」では、男性の約3分の1で認められるという報告があります。女性で目の症状が見られることは、非常にまれと考えられています。
その他、非常にまれな合併症として、びまん性平滑筋腫という種類の良性腫瘍が見られます。食道に最もよく見られ、女性生殖器や気管にも見られることがあります。
これらの症状は、必ずしも全てが同時に認められるわけではありません。
| アルポート症候群で見られる症状 |
|---|
99~80%で見られる症状 びまん性糸球体基底膜菲薄化(糸球体基底膜が薄くなる) |
79~30%で見られる症状 高血圧、メサンギウム細胞増多、タンパク尿 |
29~5%で見られる症状 角膜内皮形態の異常、前部円錐水晶体、巣状分節性糸球体硬化症、補体成分C3の糸球体沈着、IgAの糸球体沈着、顕微鏡的血尿、腎炎、ネフローゼ症候群、(再発性の)角膜びらん、腎糸球体に泡沫細胞が見られる、尿細管萎縮、網膜の斑点、感音難聴、ステージ5の慢性腎臓病、糸球体基底膜の肥厚、糸球体毛細血管壁の肥厚、尿細管間質性線維症 |
4~1%で見られる症状 大動脈瘤、陰核肥大、咳、びまん性平滑筋腫症、嚥下障害、呼吸困難、上腹部痛、黄斑変性症、前囊下白内障、再発性気管支炎、喘鳴、嘔吐 |
割合は示されていないが見られる症状 先天白内障(発達白内障)、副甲状腺機能低下症、魚鱗癬、円錐水晶体、近視、腎不全、血小板減少症 |
海外の複数の報告から、アルポート症候群の発生率は4万~5万出生に1人と考えられています。日本にいる患者さんの数は、調査研究の結果から、約1,200人と推測されています。しかし、アルポート症候群の診断がついておらず「原因不明の腎不全」とされている患者さんもいる可能性があり、また、生命予後が良好な人が多いため、幅広い年齢にわたり、多くの患者さんがいる可能性があります。
女性の場合、重症度によっては妊娠・出産の際に、産科医、小児および内科の腎専門医が連携して慎重に管理されます。
アルポート症候群は、国の指定難病対象疾患(指定難病218)、および、小児慢性特定疾病の対象疾患です。
何の遺伝子が原因となるの?
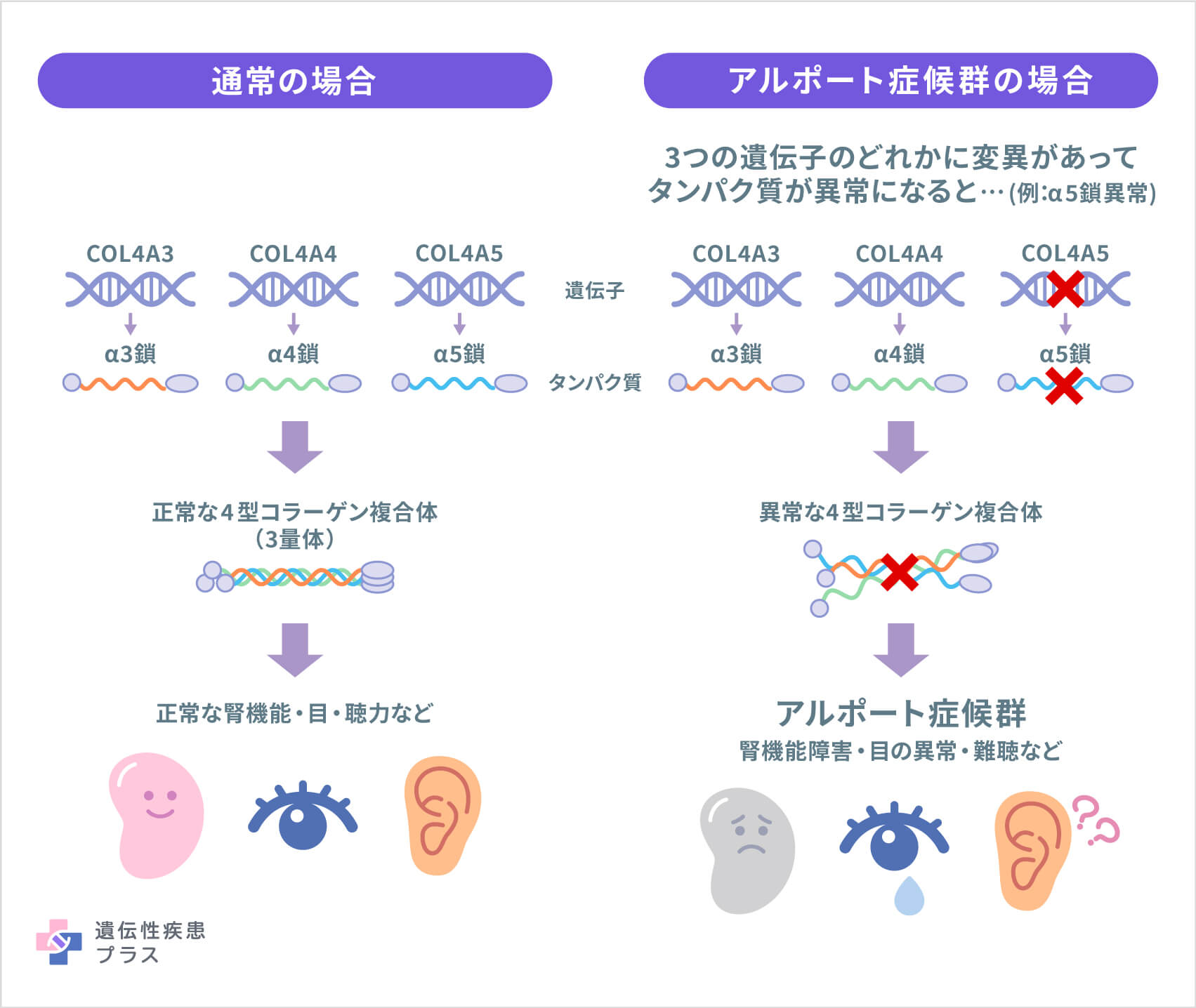
アルポート症候群の原因遺伝子として「COL4A3」「COL4A4」「COL4A5」の3つの遺伝子が見つかっています。COL4A3遺伝子とCOL4A4遺伝子は2番染色体の2q36.3という位置、COL4A5遺伝子はX染色体のXq22.3という位置に存在します。
これら3つの遺伝子は、それぞれ4型コラーゲンと呼ばれるタンパク質の構成成分(番号順にα3鎖、α4鎖、α5鎖)の設計図となる遺伝子です。4型コラーゲンは、腎臓の、特に糸球体という部分で重要な役割を果たしています。糸球体は、血液から老廃物などをろ過して尿にする、特殊な血管の集まりです。4型コラーゲンの遺伝子に変異があって糸球体が正しく作られないと、腎臓が血液を正しくろ過できなくなり、血尿やタンパク尿などの症状が現れます。
4型コラーゲンは、内耳や目にも存在します。音を聞き取る機能や、目のレンズと網膜が正しい形を保つことなどにおいて重要な働きをしているため、その遺伝子変異により、難聴や目の異常が合併する場合があります。
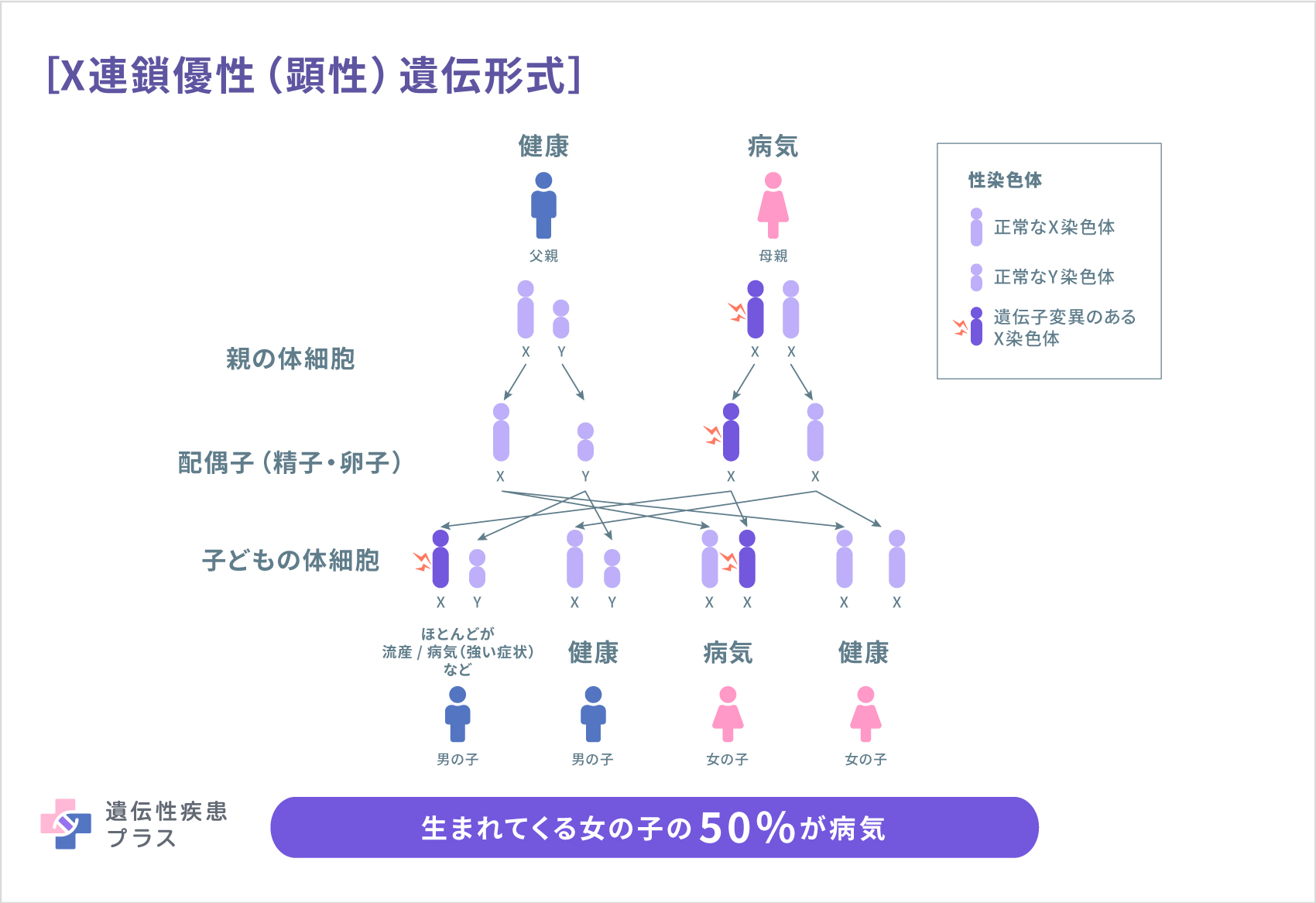
アルポート症候群は遺伝性の疾患であるため、約8割の患者さんで、家族に腎炎の人がいます。残りの約2割は家族歴がなく、子どもで新たな変異が起こり発症します。
アルポート症候群の8割は、COL4A5遺伝子変異が原因で起こります。その場合、X連鎖優性(顕性)遺伝と呼ばれる形式で遺伝します。人間は、性染色体と呼ばれる染色体を2本持っています。性染色体にはX染色体とY染色体の2種類があり、XYの組み合わせは男性、XXの組み合わせは女性になります。COL4A5遺伝子は、X染色体に存在する遺伝子です。男性が1本のみ有するX染色体のCOL4A5遺伝子に変異があると、正常なα5鎖タンパク質は全く作られないため、症状が重症になります。一方、女性の場合、X染色体を2本持っているので、1本が異常でも、もう1本のCOL4A5遺伝子が正常に働き、機能を補完します。そのため、多くの場合、男性に比べて軽症となります。しかし、一部重症化する女性がおり、これは「X染色体不活化」と呼ばれる現象のためと推測されています。
COL4A3遺伝子またはCOL4A4遺伝子の変異によるアルポート症候群は、15%程度が常染色体劣性(潜性)遺伝形式、5%程度が常染色体優性(顕性)遺伝形式で遺伝しています。これらの遺伝子変異によるアルポート症候群の場合、症状に男女差はありません。


どのように診断されるの?
アルポート症候群は、臨床所見、家族歴、腎生検や遺伝子検査の結果などから、総合的に診断が行われます。学校検診などで血尿が見つかり、それがきっかけで診断がつくことも多くあります。
日本で用いられているアルポート症候群の診断基準は、厚生労働省科学研究費補助金 難治性疾患等政策研究事業「腎・泌尿器系の希少・難治性疾患群に関する診断基準・診療ガイドラインの確立」班が作成したものです。平成27年2月改定版によると、以下の1および2を満たす、もしくは1および3を満たす場合、アルポート症候群と診断されます。
1. 主項目として「持続的血尿」が見られる
少なくとも2回検尿を行い、3か月は持続していることが確認された場合、持続的血尿とされます。アルポート症候群による腎不全が進行した状態では、まれに血尿が出なくなることもありますが、その場合には腎不全などのしかるべき兆候について確認が行われます。
2. 以下の副項目の1つ以上を満たす
- 4型コラーゲン遺伝子変異(COL4A3またはCOL4A4のホモ接合体またはヘテロ接合体変異、または、COL4A5遺伝子のヘミ接合体(男性)またはヘテロ接合体(女性)変異)
- 4型コラーゲン免疫組織化学的異常(4型コラーゲンα5鎖などに対する抗体で免疫染色を行い、染色される部位やパターンから判断される)
- 糸球体基底膜特異的電顕所見(糸球体基底膜の形状の変化などを確認して判断される)
3.以下の参考項目の2つ以上を満たす
- 腎炎・腎不全の家族歴
- 両側感音性難聴
- 特異的眼所見:前円錐水晶体(anterior lenticonus)、後嚢下白内障(posterior subcapsular cataract)、後部多形性角膜変性症(posterior polymorphous dystrophy)、斑点網膜(retinal flecks)など
- びまん性平滑筋腫症
主項目である「持続的血尿」のみが見られる場合で、家族がアルポート症候群と診断されている場合は「アルポート症候群の疑い」となります。また、副項目の「4型コラーゲン遺伝子変異」「4型コラーゲン免疫組織化学的異常」のいずれかのみが見られる場合は、無症候性の保因者と診断されます。いずれの場合も、糖尿病による腎不全の家族歴や老人性難聴など、他の疾患とわかるものは除外されます。
なお、血尿が確認された場合、原則としてすべての人で、アルポート症候群かどうかの確認(鑑別)が行われます。アルポート症候群を最も確実に診断できるのは、遺伝子検査です。しかし、遺伝子検査が可能な施設は限定されており、また、かかる費用などを考慮すると、疑いのあった場合にまず遺伝子を調べるという流れ(診断の第一選択)には、今のところはなりません。
どのような治療が行われるの?
今のところ、アルポート症候群を根本的に、つまり、遺伝子から治すような治療法は見つかっていません。そのため、末期腎不全への進行を抑えるために腎臓を保護する治療が中心となります。
具体的には、「アンジオテンシン変換酵素(ACE)阻害薬」や「アンジオテンシン受容体拮抗薬(ARB)」を内服します。腎機能障害が認められない時期には、特に生活上の制限は必要なく、通常の日常生活を送ることができます。そのため、こうした薬による治療は、血尿に加えてタンパク尿が見られるようになった頃に開始されることがほとんどです。一方で、海外では、男性の患者さんに対しては診断がついたらすぐに薬での治療を開始することを推奨するという報告もあります。
腎障害が進行して、慢性腎不全(末期腎不全)となった場合には、透析療法(血液透析、腹膜透析)や腎臓移植などが必要になります。
新薬の開発もいくつか進められています。例えば、腎機能の改善が期待できる低分子化合物「バルドキソロンメチル」は、2021年5月に「アルポート症候群における腎機能の改善」を予定される効能又は効果として厚生労働省より希少疾病用医薬品の指定を受け、同年7月には協和キリン株式会社よりアルポート症候群を対象とした製造販売承認申請が厚生労働省に行われています。また、COL4A5遺伝子の変異がある部分に「エクソンスキッピング」という現象を起こすことで、α5鎖を重症型の変異から軽症型の変異に変換させる核酸医薬の研究も進められており、まだ治験は始まっていませんが、動物実験では効果が認められています。
その他、難聴に対しては、補聴器が使用されることもあります。
どこで検査や治療が受けられるの?
日本でアルポート症候群の診療を行っていることを公開している、主な施設は以下です。
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
難病の患者さん・ご家族、支えるさまざまな立場の方々とのネットワークづくりを行っている団体は、以下です。
米国のアルポート症候群の患者支援団体(財団)で、ホームページを公開しているところは、以下です。
参考サイト
- 難病情報センター
- 小児慢性特定疾病情報センター 慢性糸球体腎炎(アルポート(Alport)症候群によるものに限る。)
- MedlinePlus
- Genetic and Rare Diseases Information Center
- Online Mendelian Inheritance in Man(R) (OMIM(R))






