



どのような病気?
ベックウィズ・ヴィーデマン症候群は臍帯ヘルニア(Exomphalos)、巨舌(Macroglossia)、巨体(Giantism)を3主徴とし、体のいろいろな部分に症状が見られる先天性の成長障害です。これらの症状の頭文字を併せてEMG症候群とも呼ばれます。
臍帯(さいたい)ヘルニアは、胎児期から肝臓、腎臓、脾臓、膵臓などの臓器の肥大がみられ、腹腔内に臓器が収まり切らずに、腸の一部がおなかからはみ出てへその緒の中に脱出している状態です。新生児期以降も鼠径(そけい)ヘルニア、臍(さい)ヘルニア、腹直筋離開などが生じて手術が必要となる場合もあります。鼠径ヘルニアは鼠径部(下腹部)の筋肉の弱い部位から腸と腹膜が筋肉の外側に出てしまうもので、脱腸とも言われます。臍ヘルニアはへその部分にヘルニアが起こったもので、いわゆるでべそと言われる状態を指します。腹直筋離開とは左右の腹直筋の間にある白線という部分が伸びて、左右の腹直筋の間が広がった状態で、腹部臓器の肥大によって生じると考えられています。
巨舌は、舌が口腔内に収まり切らない場合には、哺乳障害や咬合障害(上下の歯の噛み合わせの障害)につながることがあり、そのような場合には舌を縮小する手術が必要となる場合があります。また、ベックウィズ・ヴィーデマン症候群では、胎児期から過成長(巨体)のほか、胎盤重量増加、羊水過多、臍帯過長(へその緒が長い状態)などが見られます。
これらのほかに、新生児期の低血糖、耳たぶの線状の溝、内臓腫大、片側肥大(体の左右のどちらかが反対側に比べて大きいこと)などが見られます。男性では不妊が見られることがあります。また、約10~15%の症例でウィルムス腫瘍、肝芽腫、横紋筋肉腫などの胎児性腫瘍がみられます。
| ベックウィズ・ヴィーデマン症候群で見られる症状 |
|---|
高頻度に見られる症状 在胎不当過大(妊娠週数に対して大きい胎児)、腫瘍、高身長 |
良く見られる症状 耳たぶの形態異常(線状の溝など)、顔面中部(頬、上顎、眼窩下縁など)の形態異常、早期骨格成熟、片側肥大、脈絡膜欠如、粗な顔立ち(皮膚や皮下組織の厚さによる眉、鼻、唇、口、顎などの輪郭が不明瞭な顔立ち)、先天性横隔膜ヘルニア、腎肥大、膵外分泌不全、胎盤肥大、巨舌、下顎突出、色素性母斑、腎障害、ポートワイン母斑(単純性血管腫)、肥満、臍ヘルニア、羊水過多、早産で生まれる、大きい後頭部、眼球突出、たるんだ皮膚、絨毛膜中隔嚢胞(胎盤の中にできる嚢胞)、臍帯ヘルニア、内臓肥大、大きな口 |
しばしば見られる症状 膵臓の形態異常、心血管の形態異常、副腎皮質悪性腫瘍、副腎細胞腫大、心肥大、口蓋裂、先天性巨大尿管、停留睾丸、腹直筋離開、顔面血管腫、哺乳障害、性腺芽細胞腫、肝芽腫、肝腫大、肥大型心筋症、甲状腺機能低下症、鼠径ヘルニア、大きな泉門(新生児の頭蓋骨の境目で骨化がまだ進んでいない部分の範囲が広い状態)、巨大腸内ポリープ、平滑筋肉腫、多発性腎嚢胞、腎芽腫、腎結石、神経芽腫、神経発達遅延、神経学的発語障害、耳硬化症(アブミ骨という耳小骨の1つが固着して難聴などが生じる状態)、多血症、横紋筋肉腫、睡眠障害、膵肥大、重複尿管、尿生殖器瘻、膀胱尿管逆流症 |
極めてまれに見られる症状 キアリ奇形(小脳や脳幹の一部が脊柱管内に脱出した状態)、偽性副甲状腺機能低下症 |
ベックウィズ・ヴィーデマン症候群の有病率は新生児1万500人~1万3,700人に1人の割合とされていますが、診断されていない軽症例を含めると、実際の患者数はさらに多いと考えられています。日本の患者数は500人~1,000人とされています。ベックウィズ・ヴィーデマン症候群は小児慢性特定疾病の対象疾患です。
何の遺伝子が原因となるの?
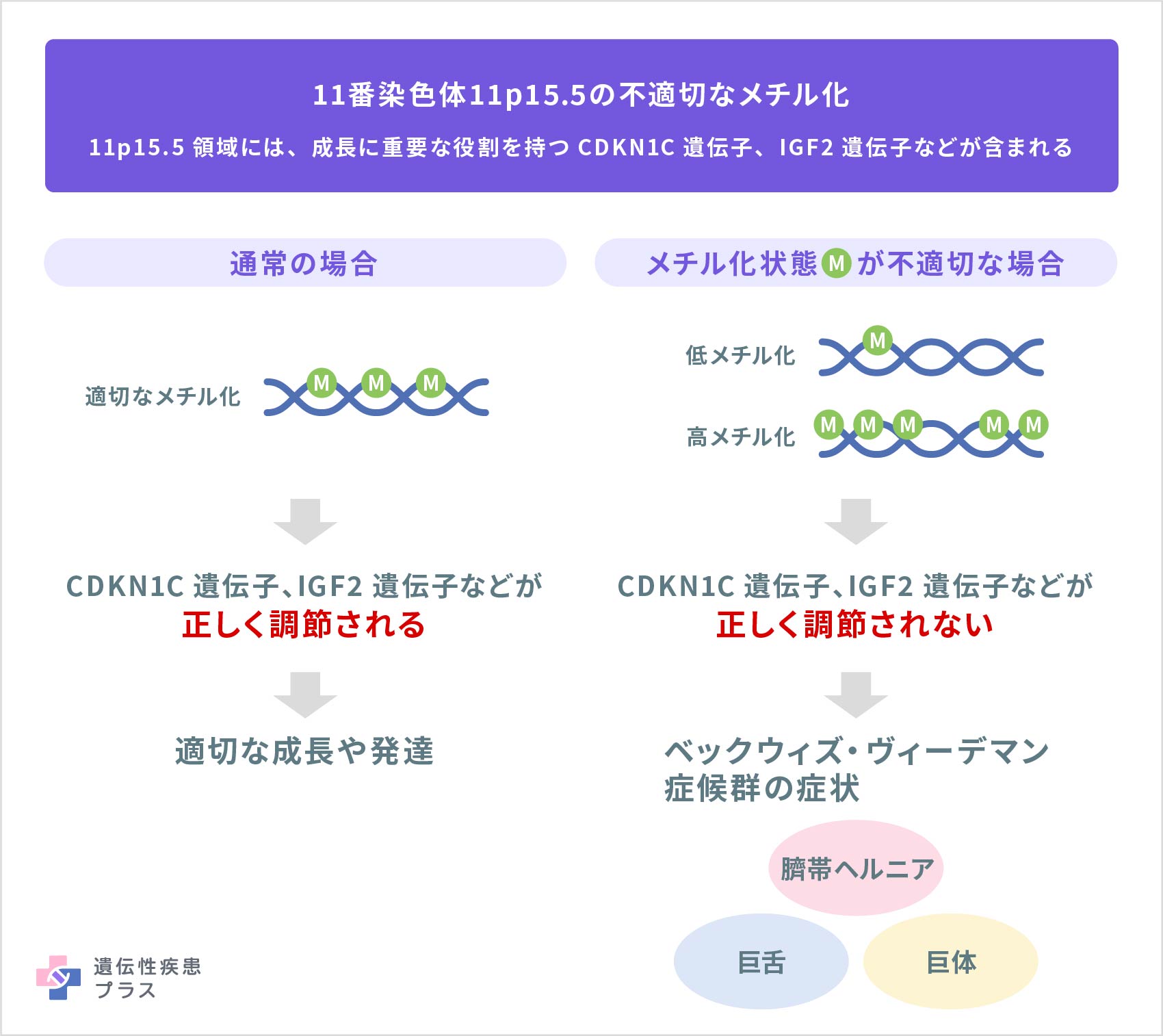
ベックウィズ・ヴィーデマン症候群の原因は複雑で、まだ完全には解明されていませんが、多くは11番染色体の11p15.5という領域にある成長に関わる遺伝子の機能の異常によると考えられています。
この領域には、ゲノムインプリンティングと呼ばれる現象が起こる遺伝子のグループが含まれています。ゲノムインプリンティングとは、両親から受け継いだどちらかの遺伝子が働かないように化学変化(主にメチル化)が起こることで、遺伝子の配列自体は変化せずに片方の親由来の遺伝子だけが働く仕組みです。ヒトの染色体は通常、父親と母親から1本ずつ受け継ぎ、ほとんどの遺伝子では、両方由来の遺伝子が細胞内で働く(発現する)状態になっています。しかし、一部の遺伝子では、父親から受け継いだものだけ、あるいは母親から受け継いだものだけが発現する必要があります。ゲノムインプリンティングは、父母由来のそれぞれ決められた特定の場所で、遺伝子の働きが調節される、重要な仕組みです。
ベックウィズ・ヴィーデマン症候群の少なくとも半数は、11p15.5領域にある、ゲノムインプリンティングを受ける遺伝子のメチル化状態の変化に関連していると知られています。そうした変化を受ける遺伝子には、CDKN1C遺伝子(KIP2遺伝子)、H19遺伝子、IGF2遺伝子、KCNQ1OT1遺伝子など、正常な成長に関与するいくつかの遺伝子が含まれています。特に、ベックウィズ・ヴィーデマン症候群の30~50%ではCDKN1C遺伝子の働きに関わる領域メチル化状態が足りないこと(低メチル化)によってCDKN1Cタンパク質の発現量が低下しており、5~10%ではIGF2の働きに関わる領域のメチル化状態が過剰なこと(高メチル化)によってIGF2タンパク質の発現量が増加していることがわかっています。これらの成長に関わるタンパク質の発現の異常が、過成長など、ベックウィズ・ヴィーデマン症候群に特徴的な症状の発症に関与すると考えられています。
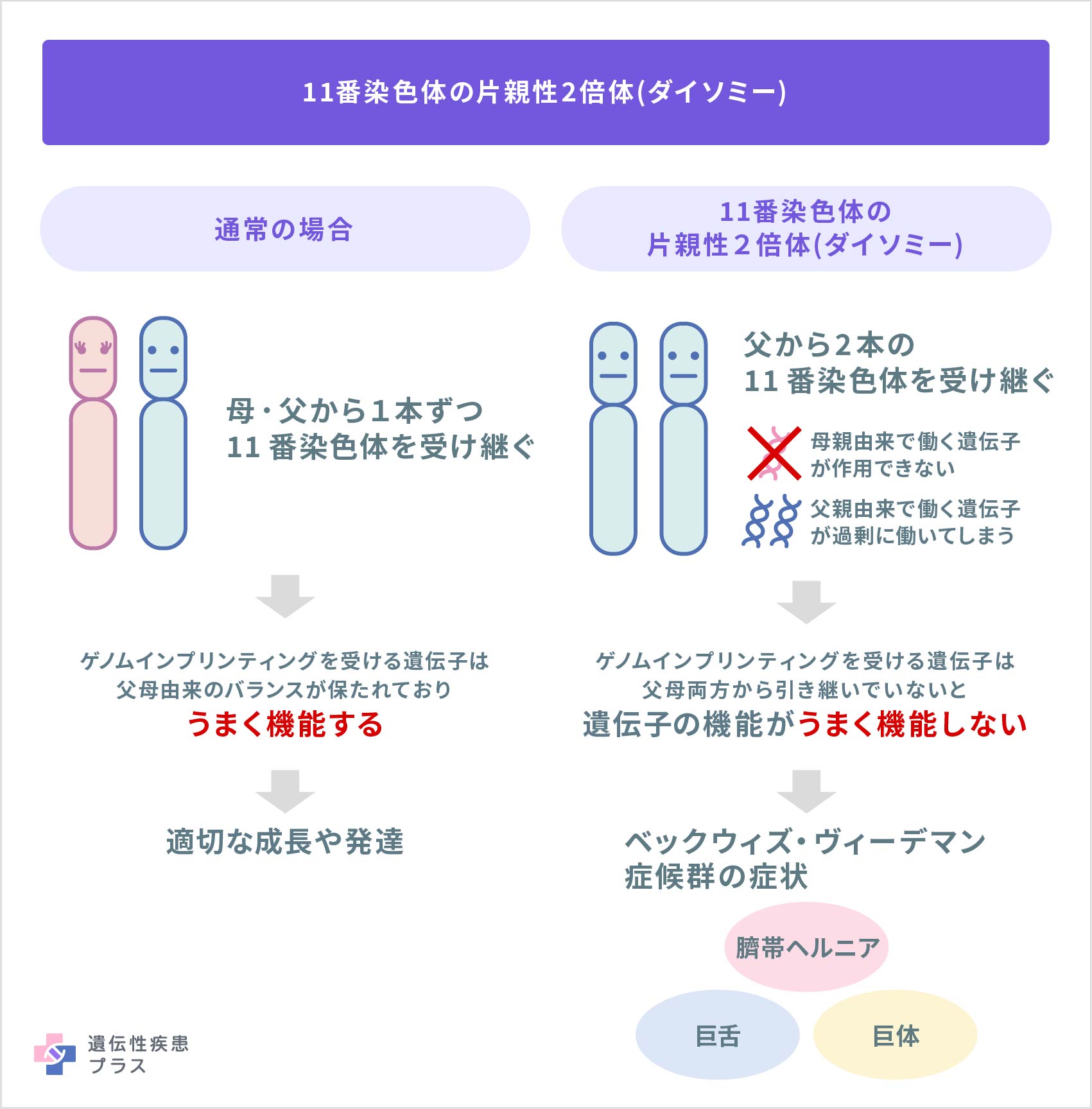
また、11番染色体に父親性2倍体と呼ばれるタイプの片親性2倍体(UPD)が起こることは、症例の約17%の原因として知られます。父親性2倍体とは、ある染色体において、何らかの理由でぞれぞれの親から1本ずつ引き継がれるのではなく、父親だけから両方のコピーを引き継いだ状態で、これも原因として知られています。11番染色体領域において、ゲノムインプリンティングを受ける遺伝子は、父母由来に応じて正しく調節されている必要がありますが、UPDによって父親由来の2つの働く遺伝子コピー、もしくは2つの働かない遺伝子コピーをもつことになり、遺伝子調節のバランスが崩れ、病気の発症を引き起こすと考えられています。
ベックウィズ・ヴィーデマン症候群の発症に関わるその他の原因として、CDKN1C遺伝子の変異(約7%)が知られています。この遺伝子は、出生前の成長を制御するところで働いている遺伝子で、変異により作られるタンパク質が変化すると、成長の抑制が妨げられ、ベックウィズ・ヴィーデマン症候群に特徴的な症状が現れると考えられています。他には、11番染色体の構造的な変化(転座、重複、欠失)が原因で発症する場合もあります(約6~10%)。
なお、全体の約3分の1の患者さんでは、遺伝子の異常が特定されていません。
ベックウィズ・ヴィーデマン症候群の大部分の患者さんは孤発性で、家族内に同じ病気の人がいるケースは15%程度とされています。しかし、第1子がベックウィズ・ヴィーデマン症候群である場合には、原因によっては、第2子以降もベックウィズ・ヴィーデマン症候群を発症する場合があります。家族性(家族内に患者さんがいる場合)では、ほとんどの場合、常染色体優性(顕性)遺伝形式で遺伝します。常染色体優性(顕性)遺伝は1対2本ある遺伝子のうち1本に異常がみられると発症する遺伝形式です。原因がCKDN1C遺伝子の変異の場合、多くは母親からの遺伝によって発症しています。また、ベックウィズ・ヴィーデマン症候群の原因が11番染色体の構造異常である場合には、遺伝する可能性があります。

どのように診断されるの?
ベックウィズ・ヴィーデマン症候群は、下記の【主要臨床症状】3つ以上、または、2つ以上と【副症状】1つ以上があれば臨床診断されます。臨床診断とは確定診断となる検査結果等がない場合に、臨床的な所見を基に診断することを指します。
また、下記の部分症状に加えて、11番染色体の11p15領域を含む染色体異常ないしインプリンティング異常、もしくは原因遺伝子(CDKN1C遺伝子等)に変異を認めれば、遺伝学的にベックウィズ・ヴィーデマン症候群と確定診断されます。
【主要臨床症状】
- 巨舌
- 巨大児
- 腹壁欠損(臍帯ヘルニア、腹直筋離開、臍ヘルニア)
- 耳垂の線状溝・耳輪後縁の小窩
- 片側肥大
- 胎児性腫瘍
【副症状】
- 新生児期低血糖
- 腹腔内臓器腫大
- 腎奇形
どのような治療が行われるの?
今のところ、ベックウィズ・ヴィーデマン症候群治す根本的な治療法はまだありません。そのため、それぞれの症状に対する、対症療法が中心となります。
臍帯ヘルニアについては必要に応じて手術が行われます。海外の報告では巨舌は成長とともに相対的な大きさが改善されるケースも見られるとされていますが、舌を縮小する手術が行われる場合もあります。
インスリンの過剰分泌による低血糖は、ベックウィズ・ヴィーデマン症候群の30~60%程度にみられますが、多くは一過性で数日~数週間で回復します。この間は血糖値が50 mg/dL以下にならないように6時間ごとに血糖値を検査して低血糖であればブドウ糖が投与されます。低血糖により脳障害を生じると、長期的加療が必要となります。
先天性心疾患で、心肥大、動脈管開存症、心房中隔・心室中間欠損症などのうち、非重篤なものについては自然治癒に至るまで超音波検査によって経過が観察されます。また、重篤な心疾患については手術が行われます。
胎児性腫瘍については、早期発見・早期治療に向けて定期的に超音波、CT、MRI等によるスクリーニング検査が実施されます。腫瘍が進展した場合には、必要に応じて化学療法や外科的手術などが行われます。また、半身肥大がある場合には、脚の長さに左右差が生じることがあるので、これを調整するための手術も考慮されます。
どこで検査や治療が受けられるの?
日本でベックウィズ・ヴィーデマン症候群の診療を行っていることを公開している、主な施設は以下です。
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
難病の患者さん・ご家族、支えるさまざまな立場の方々とのネットワークづくりを行っている団体は、以下です。
参考サイト
- 難病情報センター
- 小児慢性特定疾病情報センター ベックウィズ・ヴィーデマン(Beckwith-Wiedemann)症候群
- MedlinePlus
- Genetic and Rare Diseases Information Center
- F. Brioude et al. Clinical and molecular diagnosis, screening and management of Beckwith–Wiedemann syndrome: an international consensus statement. Nature Reviews Endocrinology 14, 229–249 (2018).






