



どのような病気?
シャルコー・マリー・トゥース病(CMT)は、末梢神経に損傷が起こり、手足の筋力低下や感覚の低下などが起こる「末梢神経疾患」の総称です。末梢神経は、脳や脊髄などの中枢神経から分かれて全身に分布する神経で、筋肉や、触覚、痛み、熱、音などの感覚を得るための感覚細胞につながっています。末梢神経の損傷は時が経つにつれて悪化し、感覚が変化または失われるようになったり、手足の筋肉の萎縮を引き起こしたりする可能性があります。
CMTでは通常、最初の症状として手足(四肢遠位部)の筋委縮がゆっくりと進行していきます。これにより、感覚が少し鈍くなるほか、足や足の指の変形、足の筋力低下により曲げたりかかとを付けたりするのが難しくなるためにスリッパが脱げやすくなったり段差につまずきやすくなったりする、「鶏歩(けいほ)」と呼ばれる特徴的な歩き方をする(両方の太ももを大きめに上げてつま先を下げて歩く)、などで気付かれるケースが多くあります。足の筋力低下や変形が原因で、足首をねんざしたり骨折したりする人もいます。しかし、国の調査により、足の問題で車いすを使用する人は約20%で、多くの人は自力歩行または杖歩行が可能だとわかっています。また、車いすのみの生活になる人はまれです。手の症状については、物を書いたり、ボタンをかけたり、ドアノブを回したりといった日常生活が行いづらくなる可能性があります。CMTでは、手足の触覚、熱、寒さなどの感覚が弱まりますが、痛みや灼熱感を感じるケースもあります。また、手足の感覚だけでなく、網膜や聴神経の障害により、目が見えにくくなったり音が聞こえにくくなったりする人もいます。その他、病気の進行とともに背骨が変形するなど、さまざまな症状が見られる人もいます。
CMTの99~80%で見られる症状 のどの異常、声の異常、運動失調、遠位筋萎縮症、筋電図の異常(神経伝導速度の低下)、歩行障害、痛みの感覚の障害、脊柱後湾症、喉頭軟化症、まひ、腱反射の減少 |
発症年齢は、一般的に0~20歳頃までですが、60歳以降に発症する人もいます。思春期~成人初期に診断される人が多くいますが、症状が強い人は、生まれてすぐに症状が出ている場合や、幼少期に発症する場合があります。大人になってから発症した人も、「子どもの頃から走るのが遅かった」「子どもの頃から足が小さかった」など、実は子どもの頃から軽い症状が出現していた人もいます。
CMTは、原因となる遺伝子異常の種類によって40以上の病型(タイプ/サブタイプ)がありますが、基本的に男女差はありません。また、同じ家族の中でも重症度や発症年齢は異なります。中には、症状が非常に軽く、CMTであることに気付かない人もいますが、ほとんどの場合、中程度の身体障害があります。ごく一部の人は重症となり(国の調査によると寝たきりになる人は約1%)、ごくまれに命に関わることもありますが、ほとんどの場合、CMTは命に影響しません。また、多くの方は仕事を続けることが可能です。
CMTの病型は、神経細胞への影響や遺伝形式によって区別されています。例えば、1型(CMT1)は、神経細胞を覆い保護する「ミエリン」の異常を特徴とします。2型(CMT2)は、神経線維や軸索の異常を特徴とします。4型(CMT4)やX型(CMTX)は、遺伝形式によって他のタイプと区別されています。また、病型には例えばCMT1A、CMT1B、CMT2A、CMT4A、CMTX1などのサブタイプがあり、それぞれ遺伝的な原因が異なります。
CMTは、1886年にシャルコー氏、マリー氏、トゥース氏の3人によって報告された歴史ある病気のため、特定の症状を示すタイプに以前呼ばれていた名前が使われることもあります。たとえば、ルシー・レヴィ―(Roussy-Levy)症候群は、手足の震えが見られるCMT1Aの一種です。デジェリン・ソッタス(Dejerine-Sottas)症候群は、幼児における重度のCMTに対し時々使われ、他に3型(CMT3)と呼ばれる場合や、変異している遺伝子によってはCMT1やCMT4に分類される場合もあります。CMTX5は、ローゼンバーグ・チュトリアン(Rosenberg-Chutorian)症候群としても知られています。
CMTは、末梢神経が関与する最も一般的な遺伝性疾患であり、世界中の人で発症しています。米国国立衛生研究所が運営するMedlinePlusの情報によると、CMTの世界的な有病率は、3,300人に1人と示されています。CMTの発症頻度は、欧米の疫学調査では2,500人に1人と報告されています。米国では約15万人がCMTだと推定されています。日本でも10万人に5~40人と推定されており、国が実施した全国調査から、日本にCMTの人は2,000人程度だと推定されています。なお、CMTになりやすい体質というものは、今のところわかっていません。
CMTは、国の指定難病対象疾患(指定難病10)です。また、「遺伝性運動感覚ニューロパチー」として小児慢性特定疾病の対象疾患となっています。
何の遺伝子が原因となるの?
CMTの原因になっていると考えられている遺伝子は、60種類上見つかっており、研究が進むにつれ増え続けています。
2021年8月現在、米国国立衛生研究所が運営するMedlinePlusに掲載されているCMTの原因遺伝子および染色体領域は、以下です。
ATP1A1、ATP7A、BSCL2、DCTN1、DNM2、DNMT1、DYNC1H1、GARS1、GJB1、HINT1、HSPB1、HSPB8、IGHMBP2、KIF1B、LMNA、MFN2、MPV17、MPZ、MT-ATP6、NAGLU、PMP22、PRPS1、SETX、SPG11、SPTLC1、SURF1、TRPV4、VCP、AARS1、ABHD12、AIFM1、ARHGEF10、BAG3、CNTNAP1、COA7、COX6A1、DCTN2、DHTKD1、DNAJB2、DRP2、EGR2、FGD4、FIG4、GDAP1、GNB4、HARS1、HK1、HSPB3、INF2、JPH1、KARS1、KIF5A、LITAF、LRSAM1、MARS1、MCM3AP、MED25、MME、MORC2、MTMR2、NDRG1、NEFH、NEFL、PDK3、PLEKHG5、PRX、PTRH2、RAB7A、SBF1、SBF2、SCO2、SGPL1、SH3TC2、SIGMAR1、SLC25A46、TRIM2、WARS1、YARS1、17番染色体17p22領域(PMP22を含む領域)
これらは、手足の末梢神経の機能に関連するタンパク質の設計図となる遺伝子です。これらの遺伝子に起きた変異が、作られるタンパク質の機能にどのようなメカニズムで影響してCMTを引き起こしているかは、まだ完全には解明されていません。しかし、神経のシグナルを伝達する軸索に障害が起きたり、ミエリンを生成する特殊な細胞に影響したりすることが、その原因につながると考えられています。こうした遺伝子に変異が起きると、多くの場合、足や手などに神経のシグナルを伝える長い神経が影響を受けやすく、その結果、末梢神経細胞は足や手の筋肉を刺激したり感覚を足や手から脳に伝達したりする能力を徐々に失っていくと考えられています。
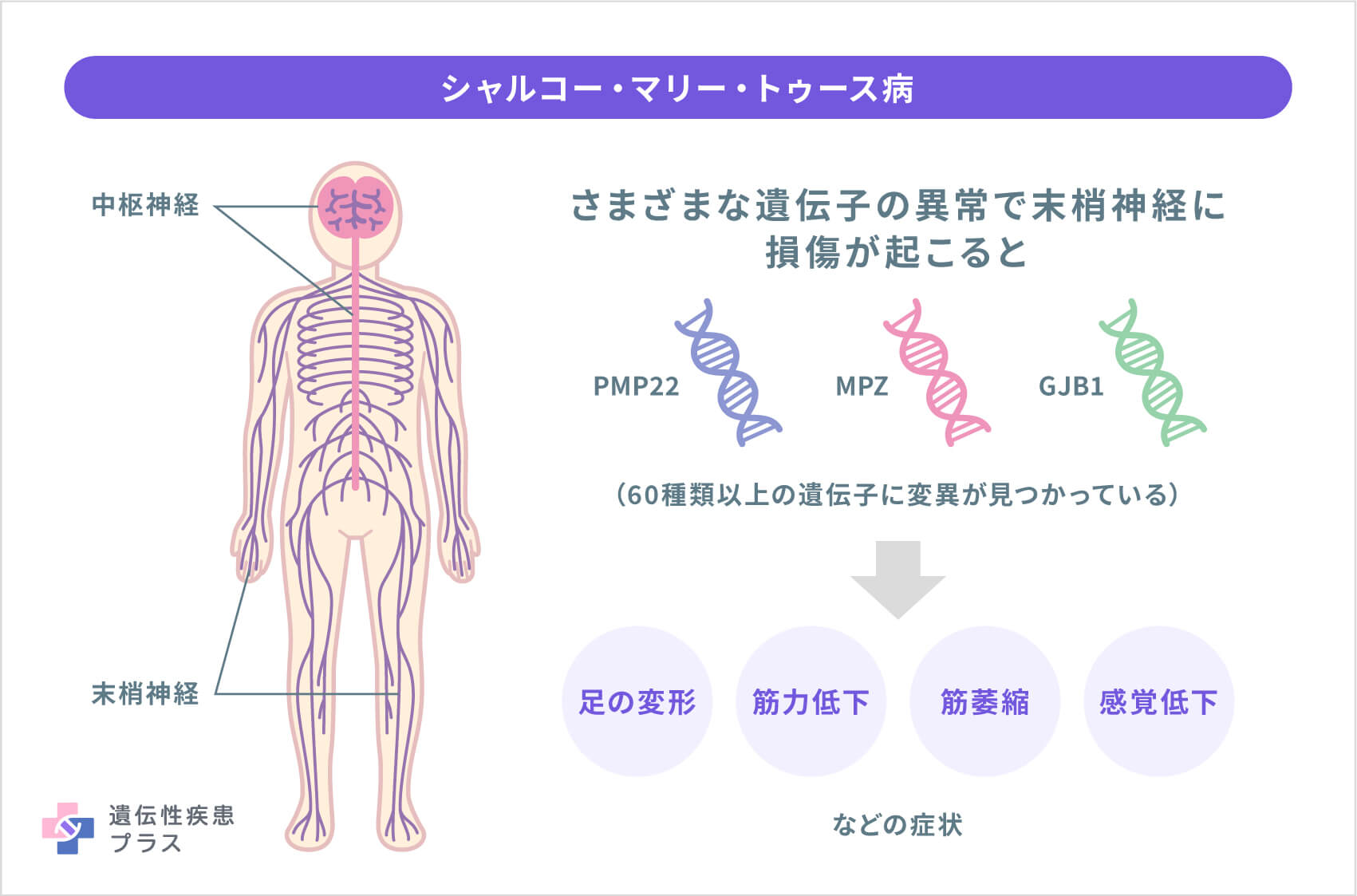
CMT1の70~80%(CMT全体の約40%)で、PMP22遺伝子に影響する変異が見られます(CMT1A型)。そのほとんどは、17番染色体の、PMP22遺伝子を含む小さな領域が重複しており、PMP22遺伝子の余分なコピーが存在しています。CMT1の残りの10~12%ではMPZ遺伝子に変異が見られます。MPZ遺伝子の変異は、CMTの他の病型でも時折認められます。CMT2では、約20%にMFN2遺伝子の変異が見られます。CMTXでは、約90%にGJB1遺伝子の変異が見られます。他の数十の遺伝子の変異は、さまざまな病型で少人数ずつ確認されています。
CMTは、変異が起きている遺伝子が異なっても、同じ症状(例えば同じような手足の筋力低下など)が見られることが、遺伝的な特徴の一つです。また、逆に、同じ遺伝子でも、変異している部分が違うと、重症度が異なったり、病型が異なったりすることがあります。
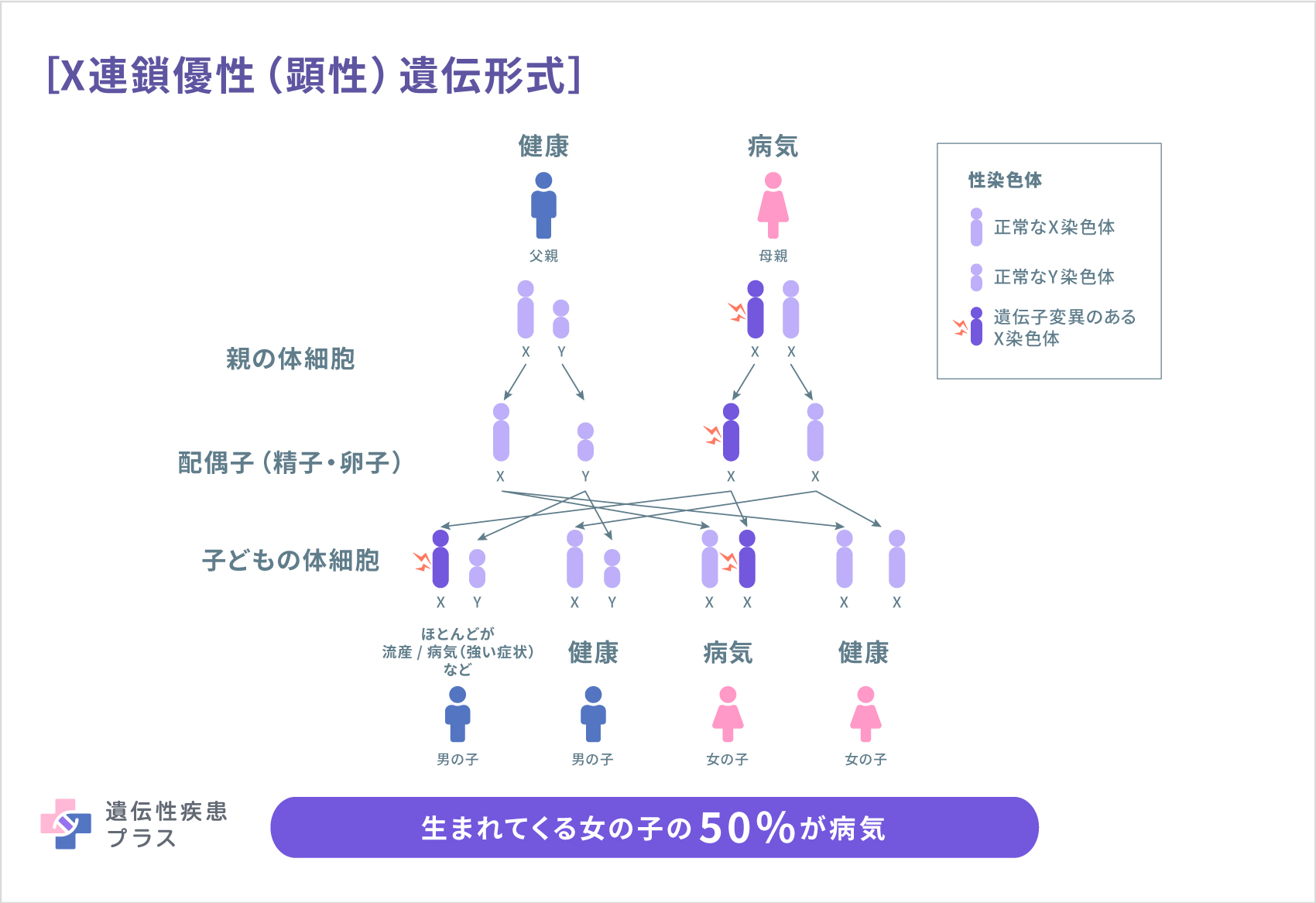
CMTの遺伝形式は、病型によって異なります。CMT1とCMT2は、ほとんどの場合、常染色体優性(顕性)形式で遺伝します。この場合、両親のどちらかがCMTで、およそ50%の確率で子どもにCMTが遺伝します。CMT4と、CMT2のいくつかのサブタイプは、常染色体劣性(潜性)形式で遺伝します。この場合、両親には症状がなくても、それぞれ変異遺伝子を1コピーずつ持つ保因者となっており、子どもでCMTが発症することがあります。CMTXはX連鎖優性(顕性)形式で遺伝します。この場合、性染色体であるX染色体上の遺伝子に変異があります。性染色体として女性はX染色体を2本(XX)、男性はX染色体を1本、Y染色体を1本(XY)、それぞれ持っています。女性でX染色体の片方に変異遺伝子が見られ、症状がある場合が、X連鎖優性(顕性)遺伝です。1本しか持たないX染色体に変異遺伝子を持つ男性は、ほとんどの場合、片方に変異遺伝子が見られて発症している女性よりも症状が強く出ます。この遺伝形式で発症した男性の子どもは、女性であれば全員片方のX染色体に変異遺伝子が見られます。その他の病型は、ほとんどが、常染色体優性(顕性)遺伝形式ですが、常染色体劣性(潜性)遺伝形式のものもいくつかあります。
常染色体優性(顕性)遺伝する病型、および、CMTXでは、両親に遺伝子変異がなくその子どもで新たに起きた遺伝子変異(新生変異、孤発例)により発症した例も複数確認されています。


どのように診断されるの?
CMTは、問診、神経学的診察、電気生理学的検査、遺伝子検査で診断されます。問診と神経学的診察でCMTが疑われた場合には、末梢神経の働きを調べる神経伝導検査が行われます。必要に応じて、針筋電図検査、神経超音波検査、神経生検(足のくるぶしのところにある腓腹神経の生検)なども行われます。神経超音波検査は無痛ですが、神経伝導検査、針筋電図検査、神経生検は痛みを伴います。これらの検査で異常が見られた場合には採血による遺伝子検査が行われ、確定診断となります。遺伝子検査は今のところ、FISH法という方法で17番染色体のPMP22遺伝子の異常を調べる検査のみ、保険適用となっています。CMTの遺伝子検査は、鹿児島大学医学部神経内科などで行われています。
どのような治療が行われるの?
今のところ、CMTを根本的に、つまり、遺伝子から治すような治療法は見つかっていません。そのため、それぞれの症状に合わせて対症療法などが行われています。
例えば、足に合った靴や下肢装具など、適切なフットケアにより、機能的な改善が期待できます。また、筋力の維持などのために理学療法や適度の運動が推奨されます。手足の変形や機能障害改善のために整形外科的手術が行われる場合もあります。歩くことに影響のある病気なので、日常生活では太りすぎに注意しましょう。
今のところ、CMTに効果があると科学的に証明された治療はありませんが、新しい治療法の研究は国内外で進められています。CMTについて正しく理解し、現在の身体機能をできるだけ維持しつつ、今後の研究成果を待ちながら、希望のある毎日を送っていくことが大切です。
どこで検査や治療が受けられるの?
日本でCMTの診療を行っていることを公開している、主な施設は以下です。
- 東北大学病院
- 仙台西多賀病院
- 新潟大学脳研究所 脳神経内科
- 千葉大学大学院医学研究院 小児病態学
- 順天堂大学医学部附属順天堂医院 脳神経内科
- 国立精神・神経医療研究センターNCNP病院 筋疾患センター
- 東名古屋病院 脳神経内科
- 名古屋市立大学病院 リハビリテーション科
- 京都大学医学部附属病院 整形外科
- 京都府立医科大学附属病院 遺伝子診療部(遺伝相談室)
- 鹿児島大学大学院医歯学総合研究科 神経病学講座
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
CMTの患者会で、ホームページを公開しているところは、以下です。
参考サイト
- 難病情報センター
- 小児慢性特定疾病情報センター 遺伝性運動感覚ニューロパチー
- KEGG DISEASE: ルシー・レヴィー症候群
- MedlinePlus
- Genetic and Rare Diseases Information Center






