



どのような病気?
肥大型心筋症は、高血圧や弁膜症など、はっきりした原因がないにも関わらず心筋(心臓の筋肉)が肥大(分厚くなる)し、そのために心臓の動きが悪くなり、さまざまな症状が見られる病気です。肥大型心筋症は、遺伝子の変異によって起こる遺伝性疾患である場合と、家族性や遺伝子の変異がなく原因不明で起こる場合があります。
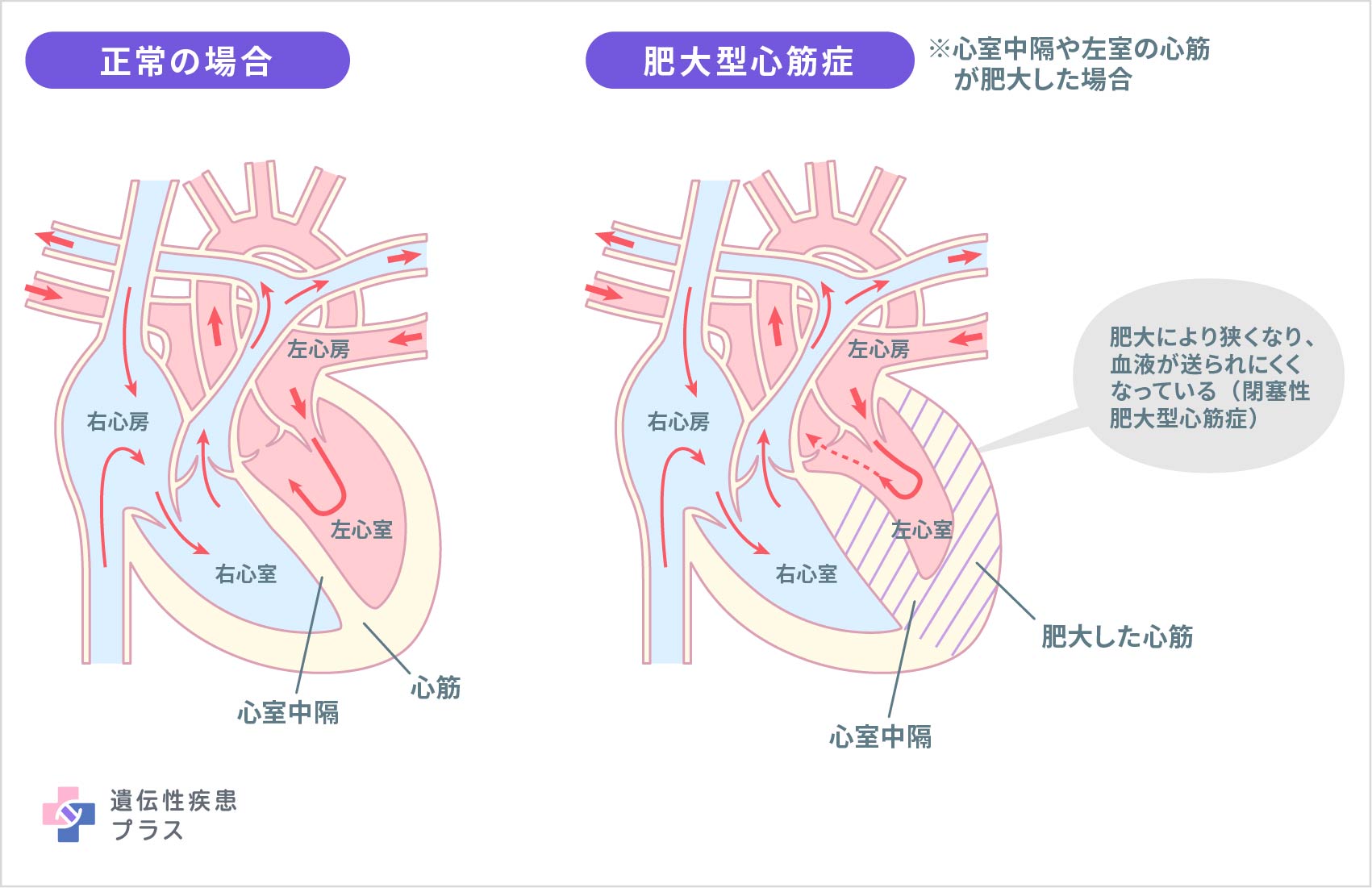
心臓は、心房と呼ばれる部分と心室と呼ばれる部分から出来ており、右の心房は右心房(右房)、左の心房は左心房(左房)、右の心室は右心室(右室)、左の心室は左心室(左室)と呼ばれています。肺から心臓に来た血液は左房に集められ、左室から全身の血管へ流されます。逆に、全身の血管から心臓に戻ってきた血液は、右房に入り、右室から肺へと流されます。このような血液の流れを作るために、心筋は収縮と拡張を絶えず繰り返しています。心筋は、心臓を取り囲む組織であると同時に、心臓の収縮機能に重要な役割を持つ筋肉です。
肥大型心筋症では、通常左室で心筋の肥大が発生しますが、右室、もしくは両方で発生することもあります。左室の心筋が肥大すると、左室の機能である左房から左室へ血液を受け入れる働きが損なわれます。特に左室と右室を隔てる心室中隔と呼ばれる部分が非対称に肥大することが最も多いとされ、左室から血液が出ていく部分に肥大がある場合、血液が出ていくための流出路が狭くなることがあり、閉塞性肥大型心筋症と呼ばれます。肥大が起こる部分は均一ではなく、さまざまな部分で起こる可能性があり、その場所や程度によって症状や重症度が異なります。閉塞性肥大型心筋症の他にも、流出路が狭くならない非閉塞性肥大型心筋症、心尖部肥大型心筋症、心室中部閉塞型心筋症、拡張相肥大型心筋症などのタイプに分かれます。
この病気の患者さんの多くは症状がほとんど見られないか、軽い症状しか見られません。そのため、検診などで心音や心電図の異常を指摘され、病気に気付く場合も多くあります。一方で、目立った症状がない場合でも、健康な人より突然死のリスクが高くなります。自覚できる症状が見られる場合、不整脈によって起こる動悸・めまい、運動時の呼吸困難・胸の圧迫感などが多いですが、閉塞性肥大型心筋症などの重症の場合には全身に血液が十分に送られなくなり、不整脈や脳虚血などが原因で失神が起こる場合もあります。
また、拡張相肥大型心筋症と呼ばれる状態は、患者さんの5~10%に発生し、左室収縮能の低下や左室拡張が進行して引き起こされます。この状態では心機能低下によって呼吸困難や動悸などのさまざまな症状が現れ、予後が悪く、心臓移植が必要となることがあります。
肥大型心筋症の発症頻度は、世界で200人に1人程度と推定されています。国内の患者さんについて、難病情報センターによれば、令和4年度末の肥大型心筋症医療受給者証所持者数は4,318人であるものの、心エコー検査スクリーニングでは500人~1,000人に1人で認められたという報告があり、診断に至っていないものの実際にはさらに多くの患者さんがいる可能性があります。
何の遺伝子が原因となるの?
肥大型心筋症は、遺伝子が原因で起こる家族性と、遺伝子が原因ではないと考えられる非家族性に分かれ、およそ半数が家族性とされています。
米ジョンズ・ホプキンス大学が運営している、ヒトの遺伝性疾患とその遺伝子のオンラインカタログOMIMには、家族性の肥大型心筋症の原因遺伝子は少なくとも26以上掲載されています。その中でも頻度が高いことが知られているのは、14番染色体の14q11領域にあるMYH7遺伝子、11番染色体の11p11.2領域にあるMYBPC3遺伝子、1番染色体の1q32.1領域にあるTNNT2遺伝子、19番染色体の19q13.42領域にあるTNNI3遺伝子です(下表参照)。
これらの遺伝子は、サルコメアと呼ばれる細胞構造に重要なタンパク質の設計図となります。サルコメアとは、筋肉収縮の基本構造であり、太いタンパク質の線維(ミオシンフィラメント)と細いタンパク質線維(アクチンフィラメント)が重なり滑り合い、互いに近づいたり離れたりすることで、フィラメントが互いに移動し、筋肉が収縮されます。
MYH7遺伝子は、サルコメアの太いフィラメントを構成する主成分となるβ-ミオシン重鎖タンパク質の設計図となり、MYBPC3遺伝子は、ミオシン結合タンパク質C(MyBP-C)と呼ばれる、太いフィラメントと結合して筋収縮の調節を助けるタンパク質の設計図となります。
TNNT2遺伝子とTNNI3遺伝子は、心筋細胞に見られるトロポニンタンパク質複合体を構成する3つのタンパク質のうちの2つである、心筋トロポニンTと心筋トロポニンIを作るための設計図となります。トロポニン複合体はサルコメアの細いフィラメントと結合し、太いフィラメントと細いフィラメントの相互作用を調節することで、筋肉の収縮と弛緩を制御する役割があります。
| 遺伝子 | 染色体上の位置 | 作られるタンパク質の主な働き | 遺伝形式 |
|---|---|---|---|
| MYH7 | 14q11 | β‐ミオシン重鎖タンパク質(ミオシンフィラメントを構成) | 常染色体優性(顕性)遺伝 |
| MYBPC3 | 11p11.2 | ミオシン結合タンパク質C(ミオシンフィラメントと結合し筋収縮調節を助ける) | 常染色体優性(顕性)遺伝 |
| TNNT2 | 1q32.1 | 心筋トロポニンT(アクチンフィラメントと結合するトロポニンタンパク質複合体を構成) | 常染色体優性(顕性)遺伝 |
| TNNI3 | 19q13.42 | 心筋トロポニンI(アクチンフィラメントと結合するトロポニンタンパク質複合体を構成) | 常染色体優性(顕性)遺伝 |
これらの遺伝子の変異によって、異常なタンパク質が作られるか、もしくは作られるタンパク質の量が減少することによってサルコメアの機能が損なわれ、正常な心筋の収縮が妨げられるのではないかと考えられていますが、これらのサルコメアに関連した遺伝子の変異が、どのようにして心筋肥大などの問題を引き起こすのか詳細はまだわかっていません。
家族性の肥大型心筋症は、ほとんどの場合常染色体優性(顕性)遺伝形式で遺伝します。この遺伝形式では親がこの病気である場合に子どもが肥大性心筋症となる確率は50%です。また、親からの遺伝ではなく孤発例として生じる場合もあります。

非家族性の肥大型心筋症は、サルコメアに関連した特定の遺伝子の変異で引き起こされるわけではありません。遺伝的原因は不明ですが、遺伝的要因と環境的要因の両方によるものではないかと考えられています。
どのように診断されるの?
肥大型心筋症の診断には、心エコー検査や心臓MRI検査が用いられ、左室肥大の程度や分布、左室流出路狭窄の有無や程度、心機能などが調べられます。
診断基準として、
1)心エコーなどの画像診断を行い、それぞれのタイプごとに以下のような所見が認められること
- 非閉塞性肥大型心筋症:心室中隔の肥大所見、非対称性中隔肥厚(拡張期の心室中隔厚/後壁厚≧1.3)など心筋の限局性肥大やびまん性肥大
- 閉塞性肥大型心筋症:左室流出路狭窄所見、僧帽弁エコーの収縮期前方運動
- 心室中部閉塞性心筋症:左室中部狭窄所見
- 心尖部肥大型心筋症:心尖部肥大所見
- 拡張相肥大型心筋症:左室駆出率低下と左室内腔の拡張を認め、肥大型心筋症からの移行が確認されたもの
2)高血圧性心疾患などではないと診断されること(鑑別診断)が必須となります。
さらに、確定診断のために、心筋生検による所見、家族性の発症を確認、遺伝学的診断などが重要となることもあります。
どのような治療が行われるの?
肥大型心筋症では、過剰な運動を避けることが重要であり、競技スポーツなどは禁止されます。閉塞性肥大型心筋症では、運動直後に流出路狭窄の程度が強くなり失神や突然死のリスクがあるため、運動中だけでなく運動直後にも注意が必要になります。
治療薬としては、左室を広がりやすくするための交感神経β受容体遮断薬やカルシウム拮抗薬、不整脈を抑える薬、血を固まりにくくする抗凝固療法の薬剤が使用されることがあります。また、突然死の原因となる重い不整脈に対して植込み型除細動器が用いられることもあります。拡張相肥大型心筋症では、心不全の治療のために利尿薬や血管拡張薬などが用いられます。
このほか、左室流出路狭窄が重症の場合には、経皮的中隔心筋焼灼術(カテーテルによってエタノール注入して心筋を焼灼する手術)、心室筋切除術が行われることがあります。拡張相肥大型心筋症における難治性心不全では心移植が行われる場合もあります。
どこで検査や治療が受けられるの?
日本で肥大型心筋症の診療を行っていることを公開している、主な施設は以下です。
- 福島県立医科大学循環器内科学講座
- 東京大学医学部附属病院高度心不全治療センター
- 東京慈恵会医科大学附属病院小児循環器外来
- 富山大学医学部循環器内科
- 金沢大学附属病院ハートセンター
- 名古屋大学附属病院ハートチーム
- 大阪大学医学部附属病院循環器内科
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
難病の患者さん・ご家族、支えるさまざまな立場の方々とのネットワークづくりを行っている団体は、以下です。






