



どのような病気?
ヒトには、さまざまな体の仕組みを調節する、「内分泌系」と呼ばれる情報ネットワークがあります。内分泌系は、「ホルモン」と呼ばれる、血流を通して体全体の細胞や組織の機能を調節する化学伝達物質によって成り立っています。多発性内分泌腫瘍症(MEN)は、主にそのホルモンを分泌する臓器である「内分泌臓器」に腫瘍が複数発生する遺伝性疾患です。
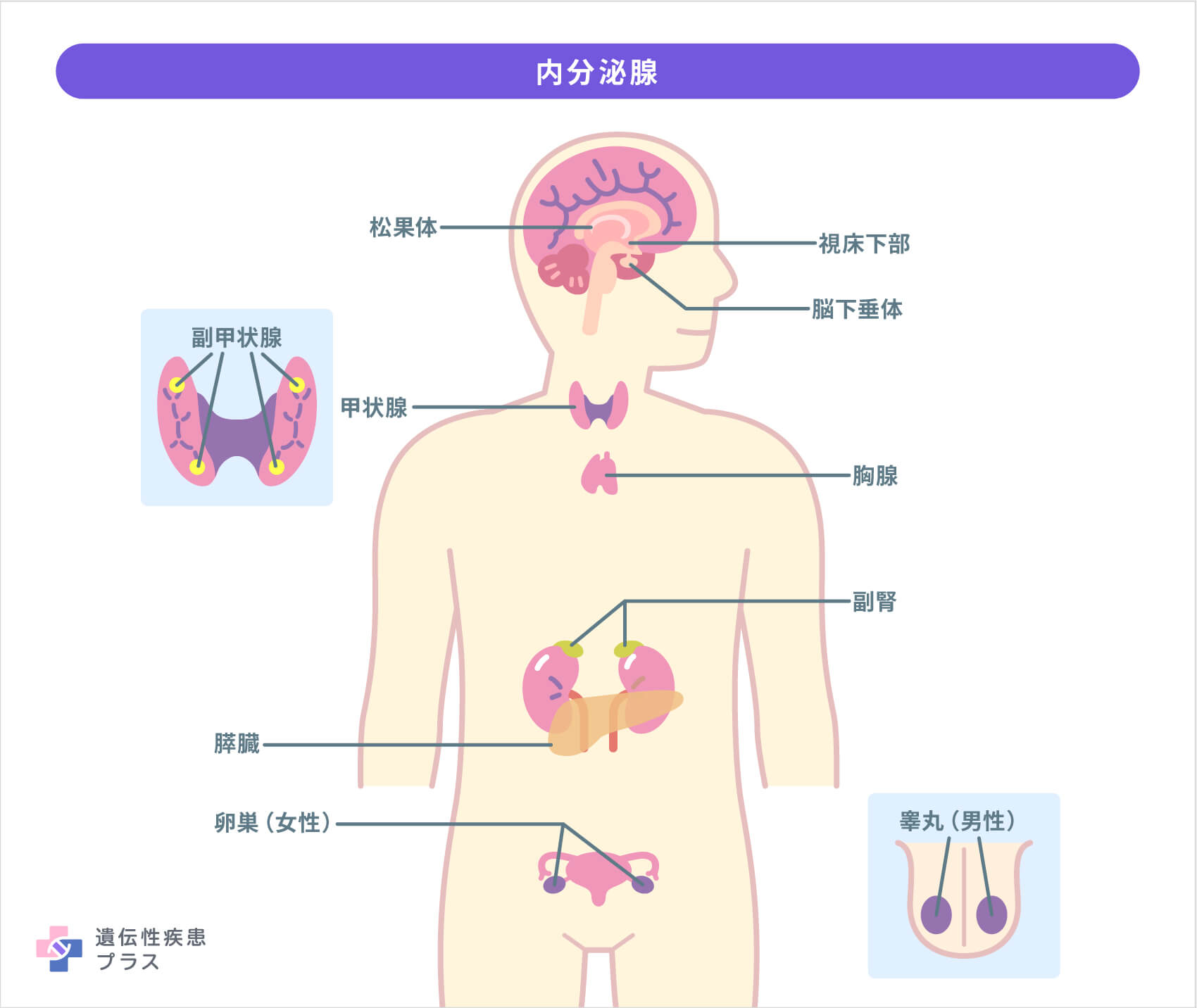
MENでは通常、少なくとも2つの内分泌腺に腫瘍が発生することに加え、内分泌臓器ではない他の臓器や組織にも腫瘍ができる可能性があります。腫瘍は良性も悪性も発生する可能性があり、悪性の場合は命に関わる場合もあります。
MENは、関与する遺伝子や作られるホルモンの種類、特徴的な臨床症状の違いによって、MEN1型、MEN2型、MEN4型の3つの型に分類されます。以前MEN3型と呼ばれていたタイプは、現在MEN2B型に分類されています。
MEN1型は、副甲状腺、下垂体、膵臓に腫瘍ができることが多くあります。この3か所すべてに腫瘍が見られるのは、MEN1型の人の2割程度です。これらの腺に腫瘍ができると、ホルモンの過剰産生につながる可能性があります。MEN 1型で最も一般的に見られるのは、副甲状腺機能亢進症です。副甲状腺機能亢進症では、副甲状腺ホルモンが過剰に分泌されることで、血液中のカルシウムの正常なバランスが崩れます(高カルシウム血症)。その影響で、腎臓結石、骨の菲薄化、吐き気と嘔吐、高血圧、脱力感、倦怠感などが起こる可能性があります。その他、副腎や皮膚、胸腺などにも腫瘍が発生することがあります。
| MEN1で見られる症状 |
|---|
99~80%で見られる症状 血管線維腫、高カルシウム血症、日常生活動作(ADL)の低下、副甲状腺過形成、原発性副甲状腺機能亢進症 |
79~30%で見られる症状 腹痛、副腎皮質異常、男性における性欲低下、勃起不全、乳汁漏出、胃食道逆流症、高カルシウム尿症、高ガストリン血症(ゾリンジャー・エリソン症候群)、大きくて境目が不規則なカフェ・オ・レ斑、複数の脂肪腫、消化性潰瘍、下垂体プロラクチン細胞腺腫、骨塩量・密度の低下、体重減少 |
29~5%で見られる症状 血漿アルドステロンの異常、副腎皮質がん、無月経、拒食症、紙吹雪様低色素斑、錯乱、便秘、脳神経圧迫、注意力低下、脱水、うつ病、十二指腸潰瘍、歯肉線維腫症、甲状腺腫、成長ホルモン過多、頭痛、吐血、高血圧、骨折しやすい、インスリノーマ(膵臓の腫瘍の一種)、腸管カルチノイド腫瘍、無気力、下血、髄膜腫、吐き気、嘔吐、腎臓結石、オステオライシス(骨の破壊)、下垂体成長ホルモン細胞腺腫、原発性コルチゾール過剰症、高身長、QT短縮(心臓の異常)、甲状腺がん |
4~1%で見られる症状 非定型欠神てんかん、昏睡、カルシトニン上昇、上衣腫、グルカゴノーマ(膵臓の腫瘍の一種)、血清セロトニンの上昇、副甲状腺がん、褐色細胞腫、下垂体皮質向性細胞腺腫、下垂体性腺刺激ホルモン細胞腺腫、下垂体前葉ホルモンの発現が認められない下垂体腺腫、下垂体甲状腺刺激性細胞腺腫、肺カルチノイド腫瘍、胸腺腫 |
割合は示されていないが見られる症状 甲状腺の異常、皮脂腺腫、副腎腺腫、下痢、食道炎、低血糖、血中コルチゾール値上昇、膵島細胞腺腫、副甲状腺腫、下垂体の非がん性腫瘍、プロラクチノーマ(下垂体腫瘍の一種)、皮下脂肪腫 |
MEN2型で、最も一般的に見られる腫瘍は、甲状腺髄様がんと呼ばれる甲状腺がんの一種です。このがんは、首のしこりとして発見されるまで無症状のことが多いとされています。一部の人では、副腎に褐色細胞腫という腫瘍ができ、これが発作性の危険な高血圧を引き起こす可能性があります。MEN2型は、「2A型」「2B型(以前は3型と呼ばれていた)」「家族性甲状腺髄様がん(FMTC)」の3つのサブタイプに分類されます。これらのサブタイプは、特徴的な症状や、特定の腫瘍が発生するリスクが異なります。こうしたMEN2型の徴候や症状は、その家族内で比較的同じように見られます。
2A型は、MENの中で最もよく見られ、ほとんどの人で甲状腺髄様がんが見られます。副腎の褐色細胞腫も約6割の人で見られます。1~2割の人で見られる副甲状腺機能亢進症は、2A型でのみ見られる症状です。
2B型は、2A型と同じく甲状腺髄様がんや褐色細胞腫が見られますが、副甲状腺機能亢進症は見られません。また、まぶた、唇、舌に粘膜神経腫が多発します。マルファン症候群に似た体型とされる、やせ形で手足が長い体型も特徴です。
FMTCでは、甲状腺髄様がんだけが見られ、その他の症状は見られません。
| MEN2で見られる症状 |
|---|
99~80%で見られる症状 甲状腺髄様がん |
79~30%で見られる症状 不安、子宮頸部腫瘍、下痢、カルシトニン上昇、尿中エピネフリン上昇、尿中バニリルマンデル酸上昇、頭痛、多汗症、褐色細胞腫に関連した高血圧、高血圧クリーゼ、顔面蒼白、動悸、副甲状腺過形成、褐色細胞腫、甲状腺C細胞過形成、甲状腺結節 |
29~5%で見られる症状 腹部膨満、舌の形態異常、巨大結腸症、頸部リンパ節腫脹、便秘、皮膚アミロイド苔癬、バランスの悪い高身長、血中甲状腺ホルモンレベルの上昇、神経節細胞腫、高カルシウム血症、高カルシウム尿症、腰椎過前弯症、後側弯症、複数の粘膜神経腫瘍、筋力低下、首の痛み、肝臓の腫瘍、肺の腫瘍、骨の腫瘍、腎臓結石、頭頸部の傍神経節腫、副甲状腺腺腫、原発性副甲状腺機能亢進症、角膜神経線維の突出、厚い唇 |
4~1%で見られる症状 関節弛緩、近位筋萎縮症、皮下脂肪の減少 |
MEN4型は、MEN1型と同様の徴候と症状が見られますが、別の遺伝子が原因となって起こります。最も一般的に見られるのは副甲状腺機能亢進症で、次いで下垂体腫瘍が多く見られます。その他の内分泌腺や、内分泌系以外の臓器の腫瘍も見られることがあります。
MEN1では、多くの場合、成人になってから内分泌臓器の機能亢進や腫瘍などの症状が出てきます。MEN2の体質を持つ人は、10歳代から甲状腺にがんにつながる変化が現れ始めます。MEN2Bは、口の周りの粘膜神経腫やマルファン症候群に似た体型などで、小児期に診断がつく場合が多いとされています。
MEN1型は約3万人に1人、MEN2型は約3万5,000人に1人が罹患していると考えられています。これを日本の人口に当てはめると、日本にいるMEN1型およびMEN2型の患者さんの数は、それぞれ約4,000人と推測されます。MEN2型の中で最も多いのが2A型、2番目に多いのがFMTCです。2B型は比較的まれで、MEN2型全体の約5%です。MEN4型の有病率は不明ですが、まれであるとされています。MENの発病頻度に男女差はなく、生命予後は、腫瘍の進行度に影響されます。
MEN1、MEN2は、小児慢性特定疾病の対象疾患です。
何の遺伝子が原因となるの?
MEN1型の原因遺伝子として、MEN1遺伝子が見つかっています。MEN1遺伝子は、11番染色体の11q13.1という場所に存在しており、メニン(menin)というタンパク質の設計図となる遺伝子です。メニンは腫瘍抑制因子、つまり、細胞が急に成長したり分裂したりするのを防ぐタンパク質として働いています。メニンの詳しい働きはまだ完全にわかっていませんが、DNAの複製や修復、他の遺伝子の活性の調節などに関わっている可能性が考えられています。この遺伝子に変異があり、メニンが腫瘍抑制因子として正しく働けなくなることで、MEN1型に特徴的な腫瘍が形成されます。
MEN2型の原因遺伝子として、RET遺伝子が見つかっています。RET遺伝子は、10番染色体の10q11.21という場所に存在しており、RETというタンパク質の設計図となる遺伝子です。RETは、細胞の分裂や成熟などの際に、細胞内で起こるシグナル伝達に関わっています。この遺伝子に変異があると、「分裂しなさい」などの指令が入らなくても、シグナル伝達が過剰に起こりっぱなしになるなどして、細胞が異常に成長や分裂を起こし、MEN2型に特徴的な腫瘍が形成されると考えられています。
MEN4型の原因遺伝子として、CDKN1B遺伝子が見つかっています。CDKN1B遺伝子は、12番染色体の12p13.1という場所に存在しており、p27というタンパク質の設計図となる遺伝子です。p27はメニンと同様に、細胞の成長と分裂を制御する腫瘍抑制因子の仲間です。この遺伝子に変異があると、細胞が無秩序に分裂して、MEN4型の症状が現れると考えられています。
これらの遺伝子変異によって特定の臓器に腫瘍が形成されるメカニズムについては、まだよくわかっていない部分が多く残されています。
MEN1型、MEN2型、MEN4型は通常、常染色体優性(顕性)遺伝形式で遺伝します。このほかに、一部の人では、家族歴がなく、生まれてくる過程で原因遺伝子に変異が起きてMENとなるケースがあります。

MEN1型では、通常、2つあるうち1つのMEN1遺伝子に変異を持って生まれます。常染色体優性(顕性)遺伝形式をとる多くの疾患は、1つが変異していることで症状が起こりますが、MEN1型はそれらとは異なり、1つのMEN1遺伝子変異を持って生まれた人が生きていく中で、あるとき特定の細胞でもう1つのMEN遺伝子にも変異が起き、それがきっかけで腫瘍が形成されると考えられています。そして、1つの変異を持って生まれたほとんどすべての人で、一生のうちに2つ目の変異が起こるとされています。
MEN2型と4型は、2つのうち1つの遺伝子が変異していることで症状が起こります。
どのように診断されるの?
小児慢性特定疾病情報センターに記載されている、MEN1とMEN2の診断方法は、以下です。
MEN1
次の3つのうちいずれかを満たす人がMEN1と診断されます。
- 「原発性副甲状腺機能亢進症」「膵消化管内分泌腫瘍」「下垂体腺腫」のうち2つ以上を有する
- 1.の3つの病変のうち1つを有し、一度近親者(親、子、きょうだい)にMEN1と診断された人がいる
- 1.の3つの病変のうち1つを有し、MEN1遺伝子に原因となる変異(病原性変異)が確認されている
なお、MEN1遺伝子変異が見つかっている患者さんの血縁者で、未発症の段階で受けた遺伝子診断によって変異が見つかり、まだ発症していない人は、医療者の間で専門的な観点から話をする際に「MEN1変異保有未発症者」と呼ばれます。こうした型の早期発見と経過観察により、腫瘍の早期治療が可能となります。
MEN2
MEN2は臨床所見と家族歴に基づいて、MEN2A、MEN2B、FMTCに細分されます。
次のうちいずれかを満たす人がMEN2AまたはMEN2Bと診断されます。
- 「甲状腺髄様がん」と「褐色細胞腫」を有する
- 1.の2つの病変のいずれかを有し、一度近親者(親、子、きょうだい)にMEN2と診断された人がいる
- 1.の2つの病変のいずれかを有し、RET遺伝子に原因となる変異(病原性変異)が確認されている
また、以下を満たす人がFMTCと診断されます。
- 家系内に甲状腺髄様がんを有し、かつ甲状腺髄様がん以外のMEN2関連病変を有さない人が複数いる
1人の患者さんの臨床像だけをもとにFMTCと診断されることはありません。MEN2Aでは、甲状腺髄様がん以外の病変については、必ず起こるとは言い切れない(浸透率が100%ではない)ため、血縁者数が少ない場合には、MEN2A とFMTCの厳密な区別は不可能とされています。MEN2Bは身体的な特徴からMEN2AやFMTCと区別されます。
MEN4については、(公財)神戸医療産業都市推進機構医療イノベーション推進センターが運営している「がん情報サイト」に、臨床的な特徴や遺伝子検査の結果などから診断が行われていることが記載されています。
どのような治療が行われるの?
今のところ、MENを根本的に、つまり、遺伝子から治すことで、腫瘍の発生や増殖を阻止するような治療法は見つかっていません。そのため、定期検査を行うことで病変をできるだけ早く発見し、その人の状況に合わせてホルモンの過剰状態に対する治療や、腫瘍摘出のための外科手術などが行われていきます。
MENでは病気に関係する臓器が複数にわたるため、患者さんはさまざまな定期検査を受け、多くの場合、手術を複数回繰り返すことになります。
手術に伴い各臓器の機能不全を生じることがあり、特にMEN1の膵腫瘍に対する治療では、部分切除であっても術後の糖尿病罹患リスクが高いとされています。また、MEN2における甲状腺髄様がんは早期治療(甲状腺全摘術)が望ましいとされています。甲状腺摘出後は、生涯にわたって甲状腺ホルモンの補充療法を受けることが必要になります。MEN2と診断された患者さんの子どもに対して遺伝学的検査を行い、変異を有するとわかった場合には発症前の予防的甲状腺全摘術を行なうことが推奨されていますが、長期的な便益と不利益の検討や、適切な手術時期については議論の余地があるとされています。
どこで検査や治療が受けられるの?
日本で多発性内分泌腫瘍症の診療を行っていることを公開している、主な施設は以下です。
- 札幌医科大学附属病院
- 福島県立医科大学附属病院 甲状腺・内分泌外科
- 群馬大学医学部附属病院 内分泌糖尿病内科
- 日本医科大学付属病院
- 虎の門病院
- 東京女子医科大学病院 内分泌外科
- 信州大学医学部内科学第四教室 糖尿病・内分泌代謝内科
- 名古屋大学医学部附属病院 乳腺・内分泌外科
- 隈病院
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
多発性内分泌腫瘍症の患者会で、ホームページを公開しているところは、以下です。
参考サイト
- 難病情報センター
- 小児慢性特定疾病情報センター 多発性内分泌腫瘍1型(ウェルマー(Wermer)症候群)
- 小児慢性特定疾病情報センター 多発性内分泌腫瘍2型(シップル(Sipple)症候群)
- 日本内分泌学会 多発性内分泌腫瘍症(Multiple Endocrine Neoplasia:MEN)
- MEN-Net.org 多発性内分泌腫瘍情報サイト
- がん情報サイト 内分泌および神経内分泌腫瘍の遺伝学(PDQ(R))
- MedlinePlus
- Genetic and Rare Diseases Information Center
- Online Mendelian Inheritance in Man(R) (OMIM(R))






