



どのような病気?
ポルフィリン症とは、「ヘム」を形成する物質であるポルフィリン(またはポルフィリン前駆体)が皮膚や血液、肝臓などの臓器に蓄積することによって起こる病気の総称です。ヘムは、ヘモグロビン(赤血球の構成成分で、酸素を体中に運ぶのに必要なタンパク質)などの鉄を含む物質の一部で、体の全ての臓器に不可欠な重要な分子です。ポルフィリン症は、ヘムの生成に関わる各種の酵素に異常があることで、ポルフィリンやその関連物質が体内に蓄積して発症する疾患です。
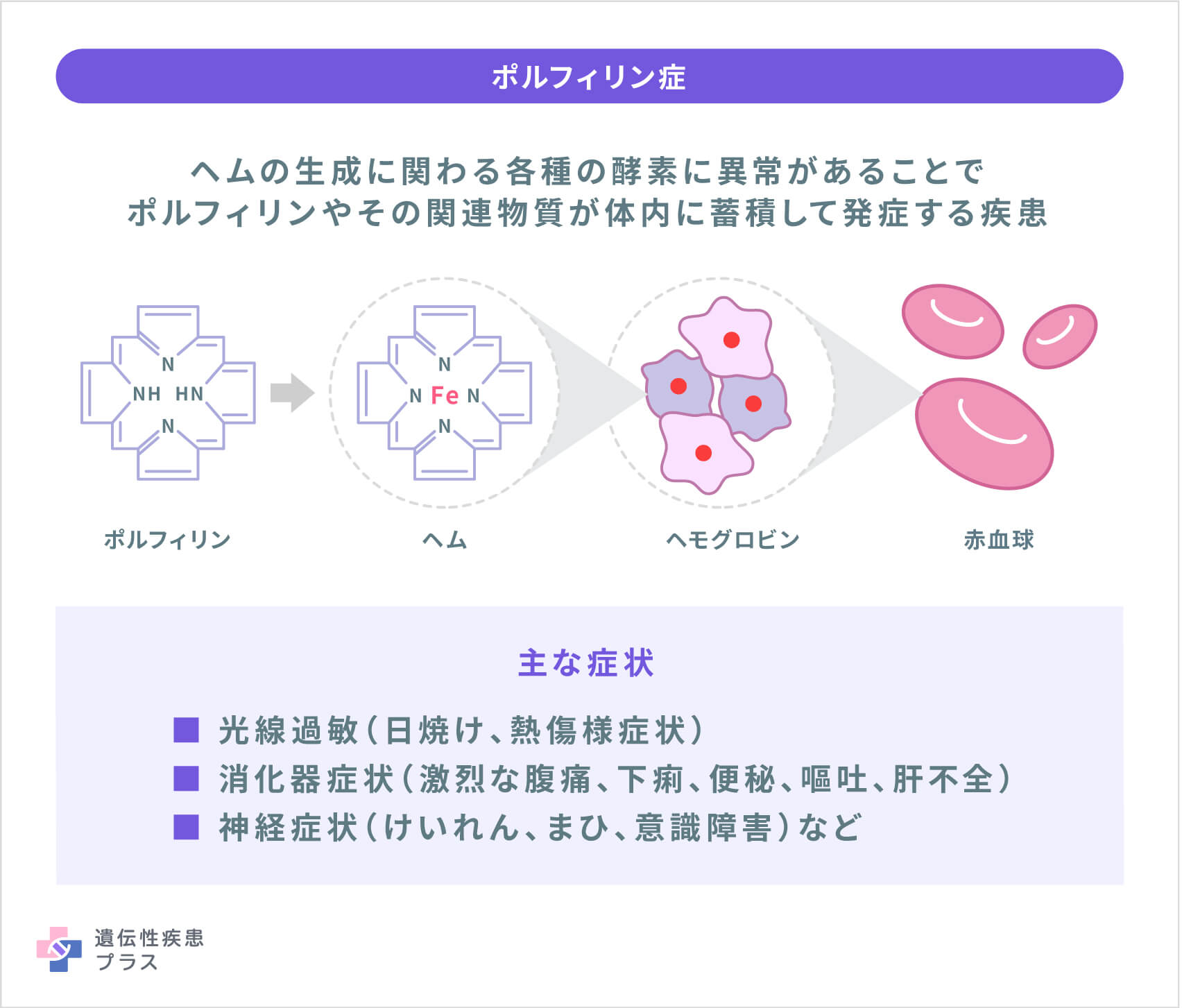
現在、ポルフィリン症には下記の9つの病型があり、光線過敏症などの皮膚症状を生じるものを「皮膚型ポルフィリン症」、腹痛、運動まひなど主に神経系に急性の症状を生じるものを「急性型ポルフィリン症」と言います。光線過敏症は日光アレルギーとも呼ばれ、日光に当たった部分の皮膚にかゆみ、発赤、炎症などが起こるものです。
ポルフィリン症は遺伝性の病気なので遺伝子の変異がある人に発症します。しかし、遺伝子変異のほかに誘因や増悪因子(喫煙、ストレス、絶食、特定の薬剤の服用など)がある場合にも症状が現れることがあると知られています。ポルフィリン症のうち、晩発性皮膚ポルフィリン症は遺伝性によることが少なく、アルコール性肝障害やC型肝炎を合併している人に発症しやすいことがわかっています。
ポルフィリン症の病型
皮膚型ポルフィリン症
| 病型 | 原因遺伝子(染色体位置) | 作られる酵素 | 遺伝形式※ |
|---|---|---|---|
| 先天性骨髄性(赤芽球性)ポルフィリン症(CEP) | UROS遺伝子(10q26.2) | ウロポルフィリノーゲンIII合成酵素 | 常染色体劣性遺伝 |
| 赤芽球性(骨髄性)プロトポルフィリン症(EPP) | FECH遺伝子(18q21.31) | フェロケラターゼ | 常染色体優性遺伝 |
| X連鎖優性(赤芽球性)プロトポルフィリン症(XLPP) | ALAS2遺伝子(Xp11.21) | δ(デルタ)アミノレブリン酸合成酵素2 | X連鎖優性遺伝 |
| 晩発性皮膚ポルフィリン症(PCT) | UROD遺伝子(1p34.1) | ウルポルフィリノーゲン脱炭酸酵素 | 常染色体優性遺伝 |
| 肝性骨髄性(赤芽球性)ポルフィリン症(HEP) | UROD遺伝子(1p34.1) | ウルポルフィリノーゲン脱炭酸酵素 | 常染色体劣性遺伝 |
急性型ポルフィリン症
| 病型 | 原因遺伝子(染色体位置) | 作られる酵素 | 遺伝形式※ |
|---|---|---|---|
| 急性間欠性ポルフィリン症(AIP) | HMBS遺伝子(11q23.3) | ポルフォビリノーゲン脱アミノ酵素(別名:ヒドロキシメチルビラン合成酵素) | 常染色体優性遺伝 |
| アミノレブリン酸脱水酵素欠損性ポルフィリン症(ADP) | ALAD遺伝子(9q32) | δ(デルタ)アミノレブリン酸脱水酵素 | 常染色体劣性遺伝 |
| 異型(多様性)ポルフィリン症(VP) | PPOX遺伝子(1q23.3) | プロトポルフィリノーゲン酸化酵素 | 常染色体優性遺伝 |
| 遺伝性コプロポルフィリン症(HCP) |
CPOX遺伝子(3q11.2) | コプロポルフィリノーゲン酸化酵素 | 常染色体優性遺伝 |
※優性遺伝は顕性遺伝、劣性遺伝は潜性遺伝とも言います。
ポルフィリン症の主な症状としては、光線過敏(日焼け、熱傷様症状)、消化器症状(激烈な腹痛、下痢、便秘、嘔吐、肝不全)、神経症状(けいれん、まひ、意識障害)などがあります。光線過敏や急性発作は生涯にわたって継続します。肝障害を伴う場合は進行すると肝硬変や肝不全となり、肝移植が必要となる場合があります。
| ポルフィリン症で見られる症状 |
|---|
99~80%で見られる症状 血中ポルフィリン濃度の異常、尿の変色 |
79~30%で見られる症状 腹痛、水疱、色素沈着、拒食症、胸痛、便秘、皮膚光線過敏症、下痢、疲労感、嘔気・嘔吐、皮膚瘙痒症、睡眠障害 |
29~5%で見られる症状 脳性まひ、発熱、幻覚、筋力低下、異常感覚、てんかん発作 |
ポルフィリン症の正確な有病率はわかっていませんが、世界の有病率は500人~5万人に1人程度と推定されています。難病情報センターに記載の情報によると、日本には約200人の患者さんがいるとされています。また、2012年までの集計で国内の患者さんは926人でした。
ポルフィリン症全体の中では晩発性皮膚ポルフィリン症が最も一般的です。また、急性間欠性ポルフィリン症も多くの国で一般的に見られ、特にスウェーデンなどの北欧諸国や英国で頻度が高い可能性があります。このように、病型によって頻度に人種差があるものもあります。
ポルフィリン症は国の指定難病対象疾患(指定難病254)および小児慢性特定疾病となっています。
何の遺伝子が原因となるの?
「どのような病気?」の表にあるように、9つの病型それぞれについて、ヘムの生成に関連する酵素に関わる遺伝子が特定されています。ポルフィリン症の発症にこれらの遺伝子変異が関与することは確実ですが、遺伝子の変異と病態の関連についてはまだ完全には解明されていません。
晩発性皮膚ポルフィリン症のうち、UROD遺伝子に変異が見られるのは約20%です。その他はUROD遺伝子変異とは関連がなく、多くの不特定な原因によって発症します(散発性)。多くの要因が晩発性皮膚ポルフィリン症の発症に寄与しています。例えば、肝臓の鉄分の増加、飲酒、喫煙、C型肝炎ウイルスやHIVの感染、特定のホルモンなどが挙げられます。ヘモクロマトーシスと呼ばれる鉄過剰症の原因となる「HFE遺伝子」の変異も晩発性皮膚ポルフィリン症と関連があるとされています。また、まだ特定されていない他の遺伝的要因も、晩発性皮膚ポルフィリン症に関連している可能性があります。
ポルフィリン症の9つの病型は、常染色体劣性(潜性)遺伝、常染色体優性(顕性)遺伝、X連鎖優性(顕性)遺伝のいずれかの形式で子どもに遺伝します。
常染色体性劣性(潜性)遺伝形式はヒトが1対2本持っている遺伝子の2本にいずれにも変異がある場合に発症するものです。両親がともに遺伝子に1本ずつ変異を有していた場合、子どもは4分の1の確率で2本とも変異を有してポルフィリン症を発症します。また、2分の1の確率で1本のみの変異を有し、発症はしない「保因者」となり、4分の1の確率で変異した遺伝子を持たずに生まれます。

常染色体優性(顕性)遺伝形式は1対2個ある遺伝子のうち1個の遺伝子の変異で発症するもので、両親のどちらかが遺伝子の変異を持つ場合に子供がその遺伝子を受け継ぐ確率は50%です。

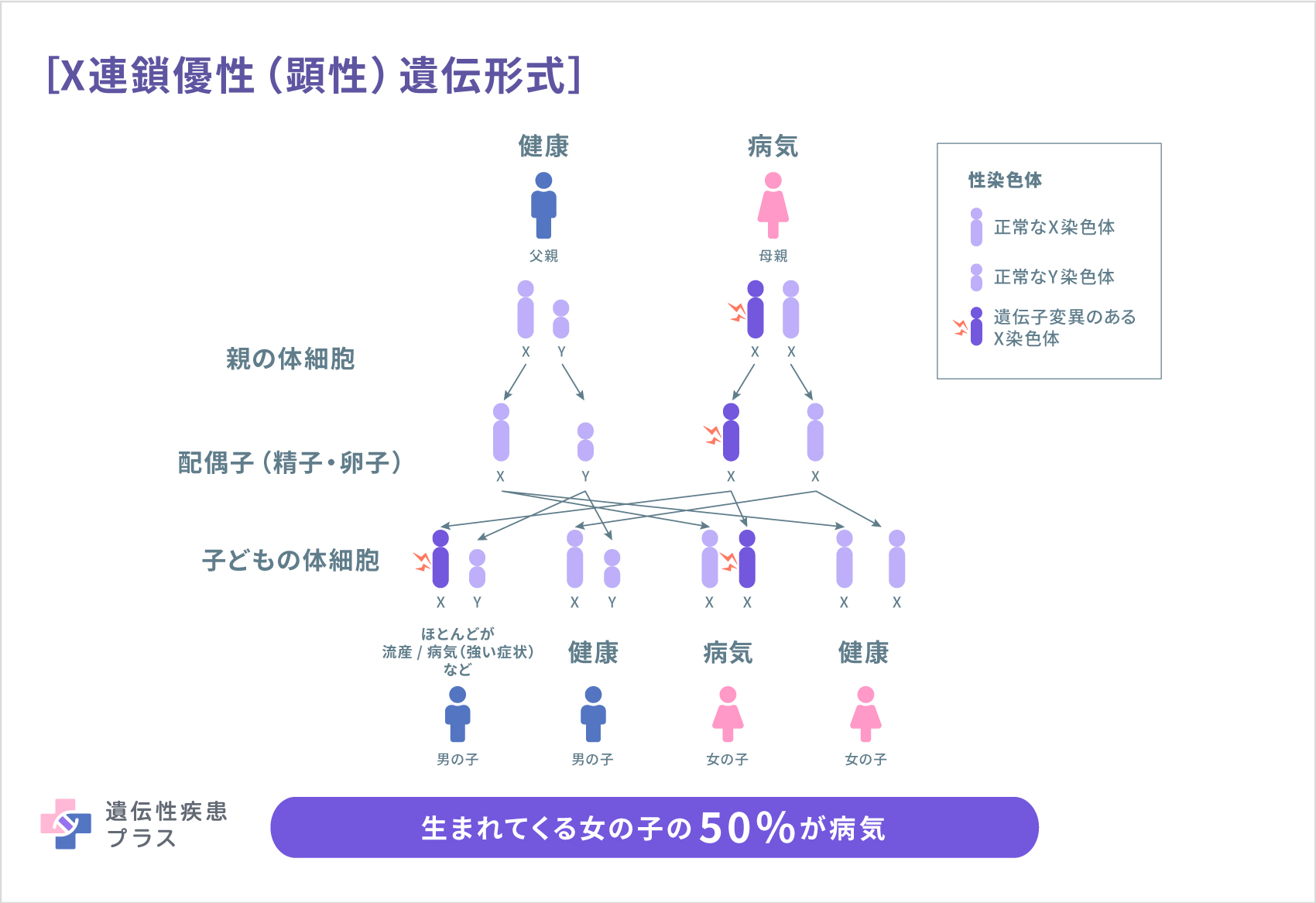
X連鎖優性(顕性)遺伝はX染色体の1個の遺伝子の異常によって発症する遺伝形式です。ヒトは、性染色体と呼ばれる染色体を1対2本持っています。性染色体にはX染色体とY染色体の2種類があり、男性はXとYの組み合わせで、女性はXとXの組み合わせになります。男性はX染色体が1本のみのため、X染色体の遺伝子に変異があると疾患が発症します。一方、女性の場合、X染色体を2本持っているので、1本が異常でも、もう1本の遺伝子が正常に働き、機能を補完するため、多くの場合、男性に比べて軽症となります。
どのように診断されるの?
ポルフィリン症は、赤血球および尿中のポルフィリン(またはポルフィリン前駆体)を測定することで、病型診断されます。いずれも、ポルフィリン症でない場合には検出されません。9つの病型について、それぞれどのようなパターンで検出されるのかは、小児慢性特定疾病情報センターの先天性ポルフィリン症・診断の手引きに、表を用いて示されています。
難病情報センターには、アミノレブリン酸脱水酵素欠損性ポルフィリン症以外の8つの病型について、診断基準が記載されています。
急性間欠性ポルフィリン症
まず、下記の【鑑別すべき疾患】に該当しないことが確認された後、以下のAまたはBのいずれかを満たす場合に急性間欠性ポルフィリン症と診断されます。
A.【臨床所見】のいずれか、および【検査所見】の①と②を満たす。
B.【臨床所見】のいずれか、および【遺伝子検査】を満たす。
【鑑別すべき疾患】
①器質的病変を基盤とする急性腹症、②イレウス(腸閉塞)、③虫垂炎、④解離性障害、⑤鉛中毒症、⑥他のポルフィリン症
【臨床所見】
①思春期以降に発症する。発症は急性のことが多い。
②種々の程度の腹痛、嘔吐、便秘(消化器症状)
③四肢脱力、けいれん、精神異常(精神神経症状)
④高血圧、頻脈、発熱など(自律神経症状)
⑤他のポルフィリン症と異なり、皮膚症状(光線過敏症)はみられない。
【検査所見(発作時)】
①尿中δ-アミノレブリン酸(ALA)の著明な増加:正常値平均値の3倍以上
②尿中ポルフォビリノーゲン(PBG)の著明な増加:正常値平均値の10倍以上
(緩解期にはALA、PBGが高値(正常上限の2倍以上)を示す)
【遺伝子検査】
ポルフォビリノーゲン脱アミノ酵素遺伝子の異常を認める。
【参考事項】
①家族歴がある
②上記症状の既往がある
③発作の誘引(バルビツール酸系・サルファ剤・抗けいれん薬・経口避妊薬・エストロゲン製剤などの薬物の服用、生理前や妊娠、出産などの性ホルモンのアンバランス、喫煙、飲酒、感染症、カロリー摂取不足、各種ストレス)がある。
遺伝性コプロポルフィリン症
まず、下記の【鑑別すべき疾患】に該当しないことが確認された後、以下のAまたはBのいずれかを満たす場合に遺伝性コプロポルフィリン症と診断されます。
A.【臨床所見】のいずれか、および【検査所見(発作時)】の①~③を全て満たす。
B.【臨床所見】のいずれか、および【遺伝子検査】を満たす。
【鑑別すべき疾患】
①器質的病変を基盤とする急性腹症(胆石発作、尿路結石など)、②イレウス(腸閉塞)、③虫垂炎、④解離性障害、⑤鉛中毒症、⑥他のポルフィリン症
【臨床所見】
①思春期以降に発症する。発症は急性のことが多い。
②種々の程度の腹痛、嘔吐、便秘(消化器症状)
③四肢脱力、けいれん、精神異常(精神神経症状)
④高血圧、頻脈、発熱など(自律神経症状)
⑤皮膚症状(光線過敏症)が見られることがある。
【検査所見(発作時)】
①尿中δ-アミノレブリン酸(ALA)の著明な増加:正常値平均値の1.5倍以上
ポルフォビリノーゲン(PBG)の著明な増加:正常値平均値の2倍以上
②尿中ウロポルフィリンの著明な増加:正常値平均値の2倍以上
コプロポルフィリンの著明な増加:正常値平均値の3倍以上
③赤血球中プロトポルフィリンは正常(ただし、緩解期には尿中ALA、PBGは正常範囲を示す)
【遺伝子検査】
コプロポルフィリノーゲン酸化酵素遺伝子の異常を認める。
【参考事項】
①家族歴がある
②上記症状の既往がある
③発作の誘引(ある種の薬物、生理前や妊娠、出産などの性ホルモンのアンバランス、喫煙、飲酒、感染症、カロリー摂取不足、各種ストレス)がある
④糞便中コプロポルフィリンの増加:正常値平均値の約360倍
異型ポルフィリン症
まず、下記の【鑑別すべき疾患】に該当しないことが確認された後、以下のAまたはBのいずれかを満たす場合に異型ポルフィリン症と診断されます。
A.【臨床所見】のいずれか、および【検査所見(発作時)】の①~③を全て満たす。
B.【臨床所見】のいずれか、および【遺伝子検査】を満たす。
【鑑別すべき疾患】
①器質的病変を基盤とする急性腹症(胆石発作、尿路結石など)、②イレウス(腸閉塞)、③虫垂炎、④解離性障害、⑤鉛中毒症、⑥他のポルフィリン症
【臨床所見】
①思春期以降に発症する。発症は急性のことが多い。
②種々の程度の腹痛、嘔吐、便秘(消化器症状)
③四肢脱力、敬礼、精神異常(精神神経症状)
④高血圧、頻脈、発熱など(自律神経症状)
⑤皮膚症状(光線過敏症)
【検査所見(発作時)】
①尿中δ-アミノレブリン酸(ALA)の著明な増加:正常値平均値の1.5倍以上
ポルホビリノゲン(PBG)の著明な増加:正常値平均値の1.5倍以上
②尿中ウロポルフィリンの著明な増加:正常値平均値の2倍以上
コプロポルフィリンの著明な増加:正常値平均値の4倍以上
③赤血球中プロトポルフィリンは正常(ただし、緩解期には尿中ALA、PBGは正常範囲内を示す)
【遺伝子検査】
プロトポルフィリノーゲン酸化酵素遺伝子の異常を認める。
【参考事項】
①家族歴がある
②上記症状の既往がある
③発作の誘引(ある種の薬物、生理前や妊娠、出産などの性ホルモンのアンバランス、喫煙、飲酒、感染症、カロリー摂取不足、各種ストレス)がある
④糞便中コプロポルフィリンの著明な増加:正常値平均値の約80倍
赤芽球性(骨髄性)プロトポルフィリン症
まず、下記の【鑑別すべき疾患】に該当しないことが確認された後、以下のAまたはBのいずれかを満たす場合に赤芽球性(骨髄性)プロトポルフィリン症と診断されます。
A.【臨床症状】および【検査所見】の①と④を満たす。
B.【臨床所見】のいずれか、および【遺伝子検査】を満たす。
【鑑別すべき疾患】
①鉄芽球性貧血症、②鉄欠乏性貧血、③溶血性貧血、④他のポルフィリン症、⑤他の光線過敏性疾患(種痘様水疱症、多形日光疹、慢性光線過敏性皮膚炎など)
【臨床症状】
光線過敏症:日光曝露後に露光部に疼痛を伴った発赤、腫脹を示す。症状が強い場合は水疱・びらんを生じる。
【臨床所見】
①皮膚症状:露光部の発赤、腫脹、顔面の虫食い状小瘢痕、色素沈着、手指関節背の苔癬化、多毛
②肝機能障害
【検査所見】
①赤血球中プロトポルフィリンの著明な増加:正常値平均値の15倍以上
②尿中ポルフィリン前駆体及びポルフィリン体は正常範囲
③赤血球蛍光が陽性
④光溶血現象が陽性
⑤露光部皮膚の血管周囲におけるPAS陽性物質の沈着
【遺伝子検査】
フェロケラターゼ遺伝子の異常を認める。
晩発性皮膚ポルフィリン症
まず、下記の【鑑別すべき疾患】に該当しないことが確認された後、以下のAまたはBのいずれかを満たす場合に晩発性皮膚ポルフィリン症と診断されます。
A.【臨床症状】および【検査所見】の①~③の全てを満たす。
B.【臨床所見】のいずれか、および【遺伝子検査】を満たす。
【鑑別すべき疾患】
①他の光線過敏性疾患(多形日光疹、慢性光線過敏性皮膚炎、光線過敏型薬疹など)、②他のポルフィリン症、③偽ポルフィリン症(臨床症状、病理所見は晩発性皮膚ポルフィリン症と類似するがポルフィリンの代謝異常を伴わない)、④多ハロゲン芳香族化合物(ダイオキシン、PCB、ヘキサクロルベンゼンなど)によるもの
【臨床症状】
光線過敏症状:日光曝露後に露光部に痒みを伴った発赤、腫脹を生じる。
【臨床所見】
①皮膚症状:露光部の発赤、水疱、びらん、露光部の瘢痕、色素沈着、多毛
②肝機能障害
【検査所見】
①尿中ウロポルフィリンの著明な増加:正常値平均値の6倍以上
ヘプタカルボキシルポルフィリンの著明な増加:正常値平均値の10倍以上
②尿中ポルフォビリノーゲン・δ‐アミノレブリン酸は正常範囲
③赤血球中プロトポルフィリン・コプロポルフィリンは正常範囲
④露光部皮膚の血管周囲におけるPAS陽性物質の沈着
【遺伝子検査】
ウロポルフィリノーゲン脱炭酸酵素遺伝子の異常を認める。
【参考事項】
①誘発の原因(飲酒、鉄材、エストロゲン製剤、経口避妊薬など)
先天性骨髄性ポルフィリン症
まず、下記の【鑑別すべき疾患】に該当しないことが確認された後、以下のAまたはBのいずれかを満たす場合に先天性骨髄性ポルフィリン症と診断されます。
A.【臨床症状】および【検査所見】の①と③を満たす。
B.【臨床所見】のいずれか、および【遺伝子検査】を満たす。
【鑑別すべき疾患】
①光線過敏性皮膚疾患:色素性乾皮症、種痘様水疱症など
②他のポルフィリン症
【臨床症状】
光線過敏症状:出生後まもなくからみられ、短時間の日光曝露により露光部に紅斑、水疱、びらん、潰瘍などを生じる。
【臨床所見】
①皮膚症状:露光部の紅斑、水疱、びらん、露光部の色素沈着、色素脱失、鼻、耳朶、手指の落屑、多毛
②赤色歯牙
③脾腫
【検査所見】
①尿中のウロポルフィリンの著明な増加:正常値平均値の35倍以上
コプロポルフィリンの著明な増加:正常値平均値の60倍以上
②血液中のコプロポルフィリンの著明な増加:正常値平均値の約30倍
③溶血性貧血
④赤血球蛍光が陽性
【遺伝子検査】
ウロポルフィリノーゲン脱炭酸酵素遺伝子の異常を認める。
X連鎖優性プロトポルフィリン症
まず、下記の【鑑別すべき疾患】に該当しないことが確認された後、以下のAまたはBのいずれかを満たす場合にX連鎖優性プロトポルフィリン症と診断されます。
A.【臨床症状】および【検査所見】の①と④を満たす。
B.【臨床所見】のいずれか、および【遺伝子検査】を満たす。
【鑑別すべき疾患】
①鉄芽球性貧血症、②鉄欠乏性貧血、③溶血性貧血、④他のポルフィリン症、⑤他の光線過敏性疾患(種痘様水疱症、多形日光疹、慢性光線過敏性皮膚炎など)
【臨床症状】
光線過敏症状:日光曝露後に露光部に疼痛を伴った発赤、腫脹を示す。症状が強い場合は水疱・びらんを生じる。
【臨床所見】
①皮膚症状:露光部の発赤、腫脹、顔面の虫食い状小瘢痕、色素沈着、手指関節背の苔癬化、多毛
②肝機能障害
【検査所見】
①赤血球中プロトポルフィリンの著明な増加:正常値平均値の15倍以上
②尿中ポルフィリン前駆体及びポルフィリン体は正常範囲
③赤血球蛍光が陽性
④光溶血現象が陽性
⑤露光部皮膚の血管周囲におけるPAS陽性物質の沈着
【遺伝子検査】
δ-アミノレブリン酸合成酵素2型遺伝子の異常を認める。
肝性骨髄性ポルフィリン症
まず、下記の【鑑別すべき疾患】に該当しないことが確認された後、以下のAまたはBのいずれかを満たす場合に肝性骨髄性ポルフィリン症と診断されます。
A.【臨床症状】および【検査所見】の①~③の全てを満たす。
B.【臨床所見】のいずれか、および【遺伝子検査】を満たす。
【鑑別すべき疾患】
①他の光線過敏性疾患(多形日光疹、慢性光線過敏性皮膚炎、光線過敏型薬疹など)
②他のポルフィリン症
③偽ポルフィリン症
④多ハロゲン芳香族化合物(ダイオキシン、PCB、へキサクロルベンゼンなど)によるもの
【臨床症状】
光線過敏症状:日光曝露後に露光部に痒みを伴った発赤、腫脹を示す。
【臨床所見】
①皮膚症状:露光部の発赤、水疱、びらん、露光部の瘢痕、色素沈着、多毛
②肝機能障害
【検査所見】
①尿中ウロポルフィリンの著明な増加:正常値平均値の6倍以上
ヘプタカルボキシルポルフィリンの著明な増加:正常値平均値の10倍以上
②尿中ポルフォビリノーゲン・δ‐アミノレブリン酸は正常範囲
③赤血球中プロトポルフィリン・コプロポルフィリンは正常範囲
④露光部皮膚の血管周囲におけるPAS陽性物質の沈着
【遺伝子検査】
ウロポルフィリノーゲン脱炭酸酵素遺伝子の病的変異をホモ接合性(1対の遺伝子の両方が変異)に認める。
【参考事項】
①誘発の原因(飲酒、鉄剤、エストロゲン製剤、経口避妊薬など)
どのような治療が行われるの?
ポルフィリン症を根本的に、つまり、遺伝子から治すような治療法はまだ見つかっていません。そのため、それぞれの症状に合わせた対症療法が行われます。
光線過敏は遮光によって予防します。光線過敏の予防に向けたサンスクリーン剤も市販されています。鉄が過剰となっている場合には瀉血(血液の一定量を取り除くこと)をして鉄の貯蔵量を低下させる治療が行われます。腹痛等の急性症状に対してはブドウ糖を含む点滴を受けます。ポルフィリン症の急性症状はポルフィリンの前駆物質が蓄積して発症します。このため急性症状に対してはポルフィリンの前駆物質の蓄積を低減するヘミン(製品名:ノーモサング点滴静注(R)250mg)という薬剤が用いられます。また、ポルフィリンの前駆物質の排泄を促進するために胆汁酸製剤やコレスチラミンという薬剤が用いられる場合もあります。
2021年8月に、急性型ポルフィリン症の4病型(急性肝性ポルフィリン症)を対象とした治療薬、ギボシランナトリウム(製品名:ギブラーリ(R)皮下注189mg)が使用開始となりました。この薬剤は、核酸医薬の一つであるRNAi治療薬という種類の薬で、アミノレブリン酸合成酵素1が作られる量を減らすことでポルフィリンの蓄積量を減らす働きをします。12歳以上の患者さんがこの薬剤を使用する場合、1か月に1回、皮下注射で投与を受けます。
急性型ポルフィリン症では服用してはいけない薬(バルビツール酸系睡眠薬、抗けいれん薬、経口避妊薬、エストロゲン製剤、サルファ剤など)がありますので、他の病気にかかった時には担当医に相談してください。また、アルコール摂取は晩発性ポルフィリン症以外の病型でも避ける方が良いとされています。
どこで検査や治療が受けられるの?
日本でポルフィリン症の診療を行っていることを公開している、主な施設は以下です。
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
ポルフィリン症の患者会で、ホームページを公開しているところは、以下です。
参考サイト
- 難病情報センター
- 小児慢性特定疾病情報センター 先天性ポルフィリン症
- MedlinePlus
- Genetic and Rare Diseases Information Center
- Online Mendelian Inheritance in Man(R) (OMIM(R))






