



どのような病気?
ソトス症候群は、大きな頭と体で生まれ、特徴的な顔立ち、成長の促進(過成長)、知的障害や運動能力の発達の遅れなどを特徴とする症候群です。筋緊張低下のため、乳児期には”はいはい”や”おすわり”が遅いなどの運動発達の遅れや、哺乳や摂食の障害もみられることがあります。乳児期にはその他、黄疸もみられることがあります。特徴的な顔立ちとして、細長い顔ととがったあご、高い額と赤い頬、目の外側の角が下がっている、などがありますが、それらの特徴は幼児期に最も顕著に現れるとされています。また、幼児期に、中耳炎や尿路感染症、けいれんなどの合併症が現れることもあります。歯も早く生えますが、過成長が目立つのは子どもの頃までで、学童期~青年期になると落ち着いてきて、多くの場合、大人になると身長は正常範囲内になります。そのほか、脊柱側弯症、てんかん発作、心臓や腎臓の障害、難聴、視力の問題なども特徴的な症状として挙げられます。また、人によって程度はさまざまですが、ほとんどの人で発達の遅れがみられます。
| ソトス症候群でみられる症状 |
|---|
99~80%でみられる症状 粗な顔立ち(顔の細かい構造がはっきりとしない)、長い腕、高身長 |
79~30%でみられる症状 骨格の成熟が早い、乱視、脳画像の異常、慢性中耳炎、便秘、頭が細長い、眼瞼裂斜下、摂食障害、顔面紅潮、全身の発達遅滞、軽度知的障害、関節が不安定、顔が長い、筋緊張低下、顔の幅が狭い、新生児黄疸が長い、突き出た額、脊柱側弯症、前髪がまばら、長いあご |
29~5%でみられる症状 攻撃的な行動、不安、大動脈瘤、脳梁の形成不全/異形成、心房中隔欠損症、自閉症様行動、てんかん発作(両側強直間代発作、ミオクローヌス発作、非運動発作)、透明中隔腔、小脳虫部形成不全、脳萎縮、計算障害、巨大大槽(脳の奇形)、局所性意識障害発作、胃食道逆流症、中等度の知的障害、重度の知的障害、脊柱後湾症、大きな手、巨頭症、動脈管開存症、足のむくみ、扁平足、協調運動不全、腎不全、震え、心室中隔欠損症、脳室拡大、膀胱尿管逆流 |
4~1%でみられる症状 足の人差し指と中指が水かきのような形でつながっている、脊椎の形態異常、急性リンパ芽球性白血病、先天性巨大結腸、足首の拘縮、星状細胞腫、注意欠如・多動性障害、両側性の屈指症、白内障、真珠腫、伝音難聴、先天性後部尿道弁、頭蓋骨癒合症、停留精巣、受精能の低下、血管腫、股関節拘縮、陰嚢水腫、水腎症、高カルシウム血症、遠視、皮膚の色素沈着過剰、部分性無歯症、皮膚の色素脱失、尿道下裂、甲状腺機能低下症、そけいヘルニア、近視、新生児低血糖症、永久歯がない、眼振、漏斗胸、包茎、肺小気胞、腎無形成、仙尾骨奇形腫、小細胞肺がん、小さな爪、斜視、内反足、臍ヘルニア、二重尿管、尿管骨盤接合部閉塞 |
割合は示されていないがみられる症状 歯が早く生える、行動異常、言語発達遅滞、前頭隆起、外反膝、耐糖能異常、前髪の生え際が高い、上あごが狭くてアーチ状になっている、反射亢進、両眼隔離症、知的障害、不釣り合いに大きい足、大きな下顎、新生児発症時の筋緊張低下、腎芽腫、中耳炎、脳梁の部分的無形成、とがったあご |
ソトス症候群の一部では、小児期にがんの発症が見られますが、特徴的に見られるがん種はなく、リスクが上がるがんもわかっていません。
ソトス症候群の患者さんは日本に約800~2,500人いるとされています。発症頻度は、1〜2万人に1人と推定されていますが、診断がついていない人たちを含めると実際の発生率は5,000人に1人に近い可能性があるとも考えられています。ソトス症候群になりやすい人種や性別はありません。また、寿命に関しては、まだ十分な情報がありませんが、60歳を超えている人もいます。
ソトス症候群は公費対象となっており、成人の場合は厚生労働省の特定疾患(指定難病194)に、18歳未満の場合は小児慢性特定疾患治療研究事業の対象疾患に指定されています。
何の遺伝子が原因となるの?
ソトス症候群の原因遺伝子として、5番染色体の5q35という位置に存在する「NSD1遺伝子」が、これまでに見つかっています。ソトス症候群患者さんの約半数は、NSD1遺伝子を含む染色体部分に微細な欠失がみられる「欠失型」で、1~2割は、NSD1遺伝子の内部に変異がみられる「変異型」です。欠失型と変異型とでは一部症状が異なるという指摘もあります。残りの3〜4割の人は、NSD1遺伝子に異常がみつかりません。
NSD1遺伝子は、「ヒストンメチルトランスフェラーゼ」と呼ばれる酵素の設計図となる遺伝子です。この酵素は、体中のさまざまな遺伝子の制御(使われ方)に関連する現象である「ヒストン修飾」に関わる遺伝子です。NSD1の制御を受ける遺伝子には、正常な成長と発達に関与する遺伝子も含まれます。NSD1遺伝子の変異により、異常なヒストンメチルトランスフェラーゼが作られ、ヒストン修飾が正しく行われなくなることが、この病気の原因と考えられています。研究では、正常なNSD1タンパク質が減少すると、成長や発達に関連する遺伝子の正常な活動が妨げられることが示されていますが、過成長や学習障害などの症状につながる仕組みは、まだ完全には解明されていません。
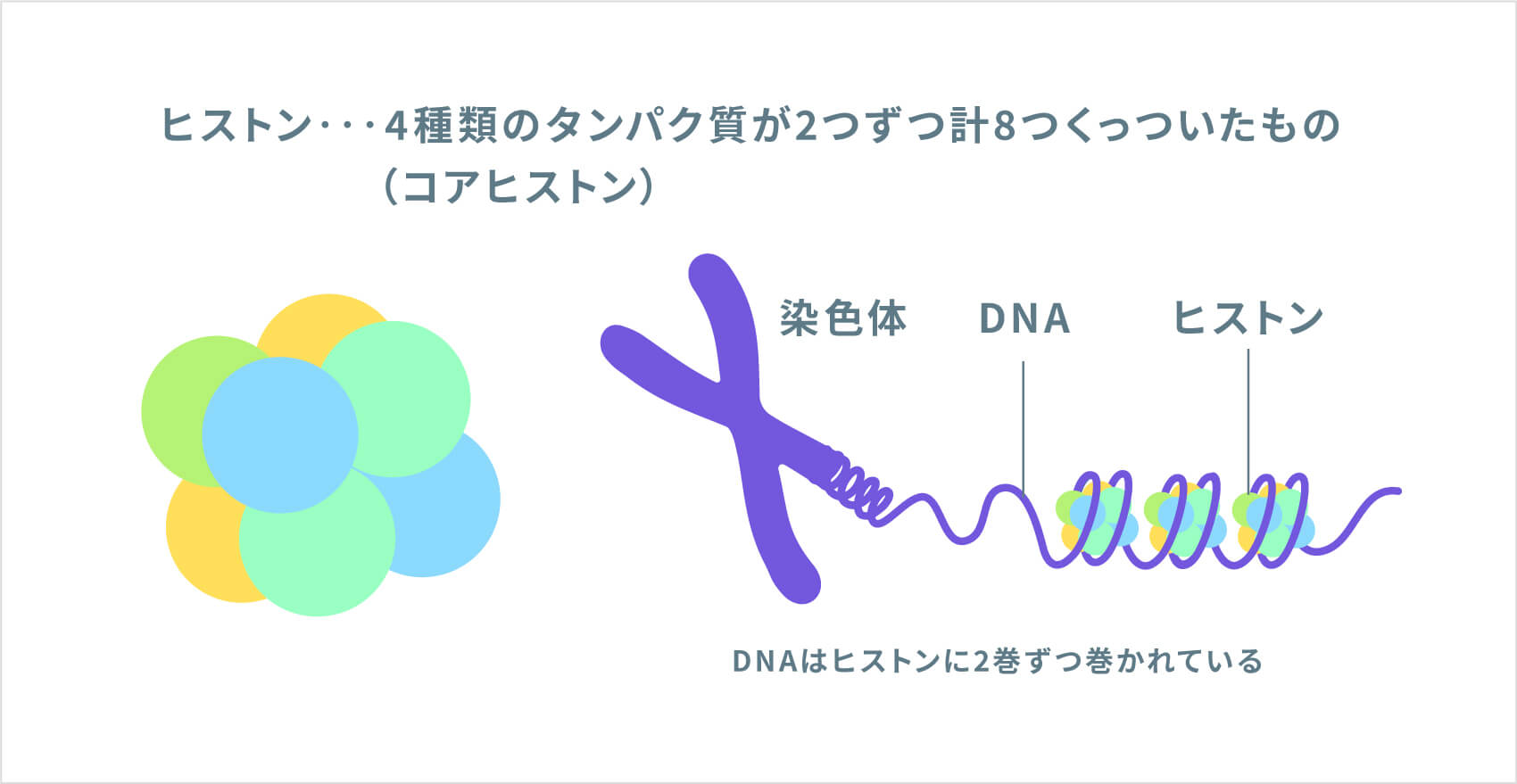
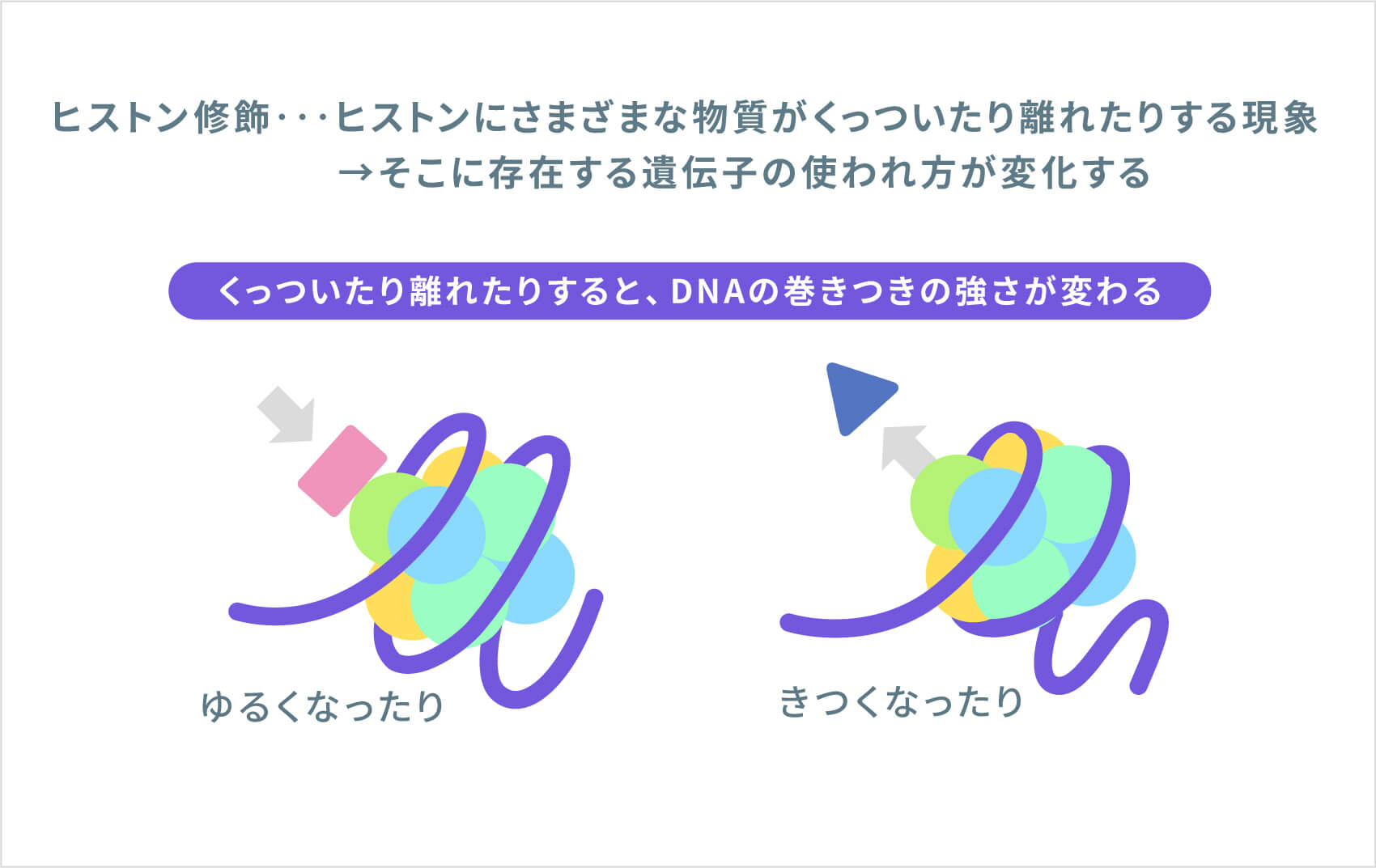

ソトス症候群の約95%は、両親には遺伝子変異がなく、子どもで新たに発生するものです(新生変異による孤発例)。残りの約5%は、ソトス症候群の親からの遺伝で発症しています。ソトス症候群の患者さんが子どもを持った場合には、常染色体優性(顕性)遺伝形式で遺伝するため、親から子へ50%の確率で遺伝します。ソトス症候群の症状や重症度は人によって異なるため、子どもがどのような症状になるかを予測することはできません。

その他、NFIX遺伝子や、APC2遺伝子の変異もソトス症候群に関連することが報告されています。これらの遺伝子はいずれも、19番染色体の19p13.13という位置に存在します。また、ソトス症候群と症状がとても似ているウィーバー症候群は、7番染色体の7q36という位置に存在するEZH2遺伝子の変異によって引き起こされることが示されています。
どのように診断されるの?
ソトス症候群には、医師がソトス症候群と診断するための「診断基準」があります。したがって、病院へ行き、必要な問診や検査を受けた後、主治医の先生がそれらの結果を診断基準に照らし合わせ、結果的にソトス症候群かそうでないかの診断をすることになります。
遺伝子を調べ、NSD1遺伝子等に変異が見つかった場合、もしくは、NSD1遺伝子を含む5番染色体長腕に欠失を認めた場合に、ソトス症候群と確定診断されます。
遺伝子に変異や欠失が認められなかった人は、次の1~4の症状を全て満たす場合に、ソトス症候群と臨床診断されます。
- 乳・幼児期の大頭症(≧2SD)
- 乳・幼児期の過成長(≧2SD)
- 頭が大きく長頭、大きい手足、前額・下顎の突出、高口蓋、眼瞼裂斜下、眼間開離を含む特徴的な外見
- 精神発達遅滞
どのような治療が行われるの?
今のところ、ソトス症候群を根本的に、つまり、遺伝子から治すような治療法は見つかっていません。そのため、それぞれの症状に合わせた対症療法が中心となります。
よりよい生活を送るために、定期的に診察を受け、合併症を早期に見つけて対応していきます。また、多くの場合、理学療法、作業療法、言語指導などの療育的支援、特別支援教育も受けていきます。
具体的には、次のような健康管理・支援が行われます。
・新生児期〜乳児期
先天性合併症の検査を行います。例えば、心エコー(心疾患の検査)、腹部エコー(腎奇形の検査)、聴覚検査(難聴の検査)、眼科診査などです。また、哺乳と摂食の支援、運動支援(理学療法)も行われます。
・幼児期
膀胱尿管逆流現象から尿路感染症を起こすことがあるため、原因不明の発熱時には尿路感染の検査もしてもらいます。中耳炎など感染症への対応、虫歯予防のための歯科健診、扁平足のチェックなども受けます。また、成長とともに今までなかった側弯が出現し、悪化してくる場合があるので、側弯のチェックも受けます。けいれんを起こす場合には、発作時の対応の仕方を医師と相談しておきましょう。その他、作業療法、言語指導など、療育への連携も行われます。
・学童期以降
身体合併症(心臓、腎臓、側弯、眼科など)の定期検査は継続的に行われます。必要に応じて、特別支援教育や、精神・心理面のケアを受けていきます。
どこで検査や治療が受けられるの?
日本でソトス症候群の診療を行っていることを公開している、主な施設は以下です。
- 東北大学病院
- 埼玉県立小児医療センター
- 国立成育医療研究センター 遺伝診療科
- 慶應義塾大学医学部 小児科
- 東京医療センター 臨床遺伝センター
- 神奈川県立こども医療センター 遺伝科
- 昭和大学横浜市北部病院 臨床遺伝・ゲノム医療センター
- 愛知県医療療育総合センター中央病院
- 大阪母子医療センター 遺伝診療科
- 長崎大学病院ゲノム診療センター 遺伝カウンセリング部門
- 医誠会国際総合病院 難病医療推進センター
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
難病の患者さん・ご家族、支えるさまざまな立場の方々とのネットワークづくりを行っている団体は、以下です。
米国のソトス症候群の患者支援団体で、ホームページを公開しているところは、以下です。
参考サイト
- 難病情報センター
- 小児慢性特定疾病情報センター ソトス(Sotos)症候群
- MedlinePlus
- Genetic and Rare Diseases Information Center
- Online Mendelian Inheritance in Man(R) (OMIM(R))






