



どのような病気?
ターナー症候群は、性染色体である「X染色体」が欠失、または一部欠失などの構造変化があることで生じる遺伝性疾患の総称です。発症するのは女性のみです。最も特徴的な症状は、低身長で、これは5歳ごろまでにはっきりしてきます。最終的な平均身長は、通常の女性より20cmほど低いと知られています。また、乳幼児期~小児期は、中耳炎になりやすく、中耳炎は繰り返すと難聴につながる恐れがあるため、注意が必要です。
性腺の機能不全も、一般的に見られます。卵巣は、初期には正常に発達しますが、卵細胞(卵母細胞)は多くの場合早期に消失し、生まれる前から卵巣組織が退化します。女性は一般的に、10~12歳ごろに、卵巣から血液中に分泌される女性ホルモンの働きにより、乳房が膨らむ、月経が始まるなどの二次性徴が起こります。ターナー症候群の女性の多くは、卵巣機能が弱いことで、女性ホルモンが正しく分泌されず、ホルモン療法を受けなければ、思春期になっても二次性徴や月経が正常に始まらず、不妊症になります。ターナー症候群の99%以上が不妊ですが、通常の不妊治療で妊娠に至ることはほとんどありません。妊娠した場合も、流産する確率が高いと知られている一方で、ターナー症候群のごく一部の人は、若年成人期まで正常な卵巣機能を維持しており、妊娠・分娩した例も報告されています。
その他、外見を含めた身体的な特徴が見られる場合もあります。具体的には、翼状頸(よくじょうけい、首の後ろの肩にかけての皮膚が広くたるむ)、首の後ろの髪の毛の生え際が低い、外反肘(がいはんちゅう、肘のところで腕が外側に曲がっている)、手足のむくみ(リンパ浮腫)、骨格の異常、腎臓や心臓の異常などです。特に成人期には、肥満・糖尿病・甲状腺ホルモン分泌低下・骨粗鬆症といった合併症が出てくる可能性があります。こうした身体的な異常が見られる人も、多くは普通の人と変わらない日常生活を送ることが十分に可能です。また、診断がついた後の定期検査をしっかり受けることで、合併症などの早期発見ができ、症状の出現や進行を食い止めることが可能な場合もあります。また、ターナー症候群の女性は、ほとんどが知的に正常ですが、まれに知的障害のある人もいます。
他にもさまざまな症状がありますが、現れる症状や表面化してくる時期、また、その程度も、人によって異なります。ターナー症候群は生まれつきの体質であり、後天的に起こることはありませんが、生まれた時点ではわからず、後にターナー症候群と診断される場合があります。
| ターナー症候群で見られる症状 |
|---|
99~80%で見られる症状 前腕の骨の形態異常、乳頭の形成不全、外反肘、思春期の遅れ、骨格成熟の遅れ、女性不妊、尿中性腺刺激ホルモン値の上昇、血清ゴナドトロピン値の上昇、恥骨を境として上半身の方が下半身よりも一般的な比率より大きい、出生前発育不全、骨減少症、骨粗しょう症、出生後の発育遅延、早発卵巣不全、短い首、低身長、短い胸骨、乳首の間隔が広い |
79~30%で見られる症状 不安、大動脈弓動脈瘤、首の幅が広い、指紋・掌紋・足底紋の異常、肝トランスアミナーゼ値の上昇、遠位大腿骨骨端拡大、乳幼児期の発育不良、外反膝、橋本病(慢性甲状腺炎)、聴覚障害、脂肪肝、アーチ形で狭い上あご、つま先関節の過可動、高血圧、足の爪の形成不全、非言語的コミュニケーション障害、近位脛骨骨端の異常、脊柱後湾症、首の後ろの髪の毛の生え際が低い、耳の位置が低い、下あごが小さい、翼状頸、肥満、原発性無月経、再発性中耳炎、下あごが後退している、続発性無月経、幅広い胸郭、手の薬指または小指の中手骨(根本側の長い骨)が短い、特定の学習障害 |
29~5%で見られる症状 爪の形態異常、歯列の異常、脱毛症、心房中隔欠損症、注意欠如・多動症、皮膚の異常な瘢痕化、二尖大動脈弁、セリアック病、胆汁うっ滞性肝疾患、大動脈の狭窄、頸部嚢胞性リンパ管腫、社会的発達の遅れ、うつ、異所性腎、内眼角贅皮、外耳の奇形、肝線維症、股関節形成不全、馬蹄腎、過度に凸化した爪、高インスリン血症、高脂血症、陥没乳頭、リンパ浮腫、マーデルング変形(手の奇形)、心筋梗塞、近視、多数の先天性色素性母斑、漏斗胸、扁平足、QT延長(心電図の異常)、眼瞼下垂、脊柱側症、つま先が短い、つま先が広がっている、斜視、2型糖尿病、白斑 |
4~1%で見られる症状 大動脈解離、動脈解離、胆汁性肝硬変、肝硬変、消化管血管形成異常、性腺芽細胞腫、左心低形成、大腸炎、悪性黒色腫、腎形成不全 |
ターナー症候群は、出生女児の約1,000~2,500人に1人が発症すると推測されており、日本には約4万人の患者さんがいます。また、流産や死産となった場合の一般的な原因の1つになっています。
ターナー症候群は、小児慢性特定疾病の対象疾患となっています。
何の遺伝子が原因となるの?
ヒトは通常、各細胞に、父由来23本、母由来23本の2セット計46本の染色体を持っています。そのうちの2本は「性染色体」で、通常女性は2本のX染色体、男性は1本のX染色体と1本のY染色体を持っています。ターナー症候群は、女性の細胞に正常なX染色体が1本存在し、もう1本は欠損している、もしくは構造的に異常になっている場合に発症します。また、正常なX染色体が1本存在し、短腕が欠けたY染色体が見られる場合(「46,XYp-」と表記されます)にも、ターナー症候群の兆候が見られます。
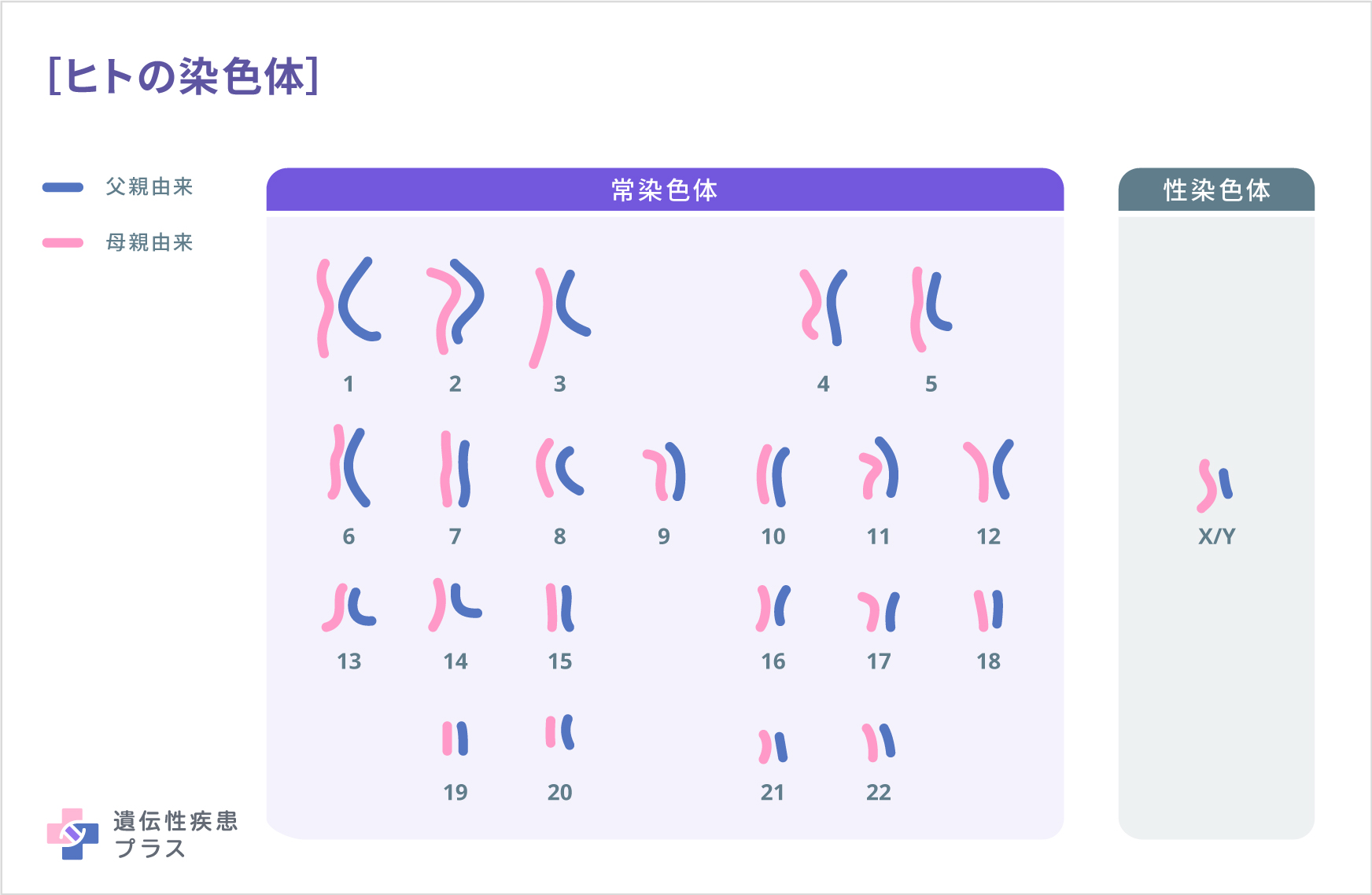
2本のX染色体のうちの1本全体がない場合、「X染色体モノソミー」といわれます。細胞の染色体が全部で46本ではなく45本になるので、「45,X」と表記されることもあります。もう1本のX染色体で見られる構造的な異常とは、短腕部分がなく、短腕があるべきところに複製された長腕がある「46,X,i(Xq)」(この状態を同腕染色体といいます)、短腕が欠けている「46,XXp-」などです。まれに、X染色体の部分的な欠失が原因でターナー症候群となる場合もあります。また、ターナー症候群の女性の中には、体全体の細胞ではなく、一部の細胞だけにX染色体の欠損や構造異常が見られる人がいます。これは「モザイク現象」として知られており、X染色体モザイクによって引き起こされるターナー症候群の女性は、「モザイク・ターナー症候群」と呼ばれます。
こうしたX染色体の数や構造の異常が、ターナー症候群で起こる症状の原因となっていると考えられていますが、発症に至るメカニズムはまだ完全には解明されていません。X染色体上の、どの遺伝子がターナー症候群の症状に関連しているかも、まだ十分にわかっていません。今のところ、X染色体短腕のXp22.23という位置に存在する「SHOX遺伝子」が、症状に関連のある遺伝子として特定されています。この遺伝子は、生まれてくるまでに体の構造を形成していく段階で働く、転写因子という種類のタンパク質の設計図となる遺伝子です。この遺伝子は、Y染色体短腕のYp11.2という位置にも存在しているため、通常、男女問わず各細胞にSHOX遺伝子が2つずつ存在します。染色体異常により、SHOX遺伝子の1つがない状態になると、ターナー症候群で見られる低身長や骨格異常が生じる可能性があることが、これまでの研究でわかっています。関連する他の遺伝子も、徐々にわかってきています。
前述のように、通常、細胞に染色体は父由来23本、母由来23本の2セット計46本あります。代々、46本の染色体を保つためには、受精前の卵子や精子では、23本1セットの染色体となる必要があります。この、23本に分かれる段階で、何らかの理由でX染色体が2本一緒に卵子もしくは精子に入ってしまい、もう一方には入らない場合があります。この現象を「染色体不分離」といいます。染色体不分離により、X染色体が入らなかった卵子/精子が受精すると、X染色体モノソミー(=ターナー症候群)となります。
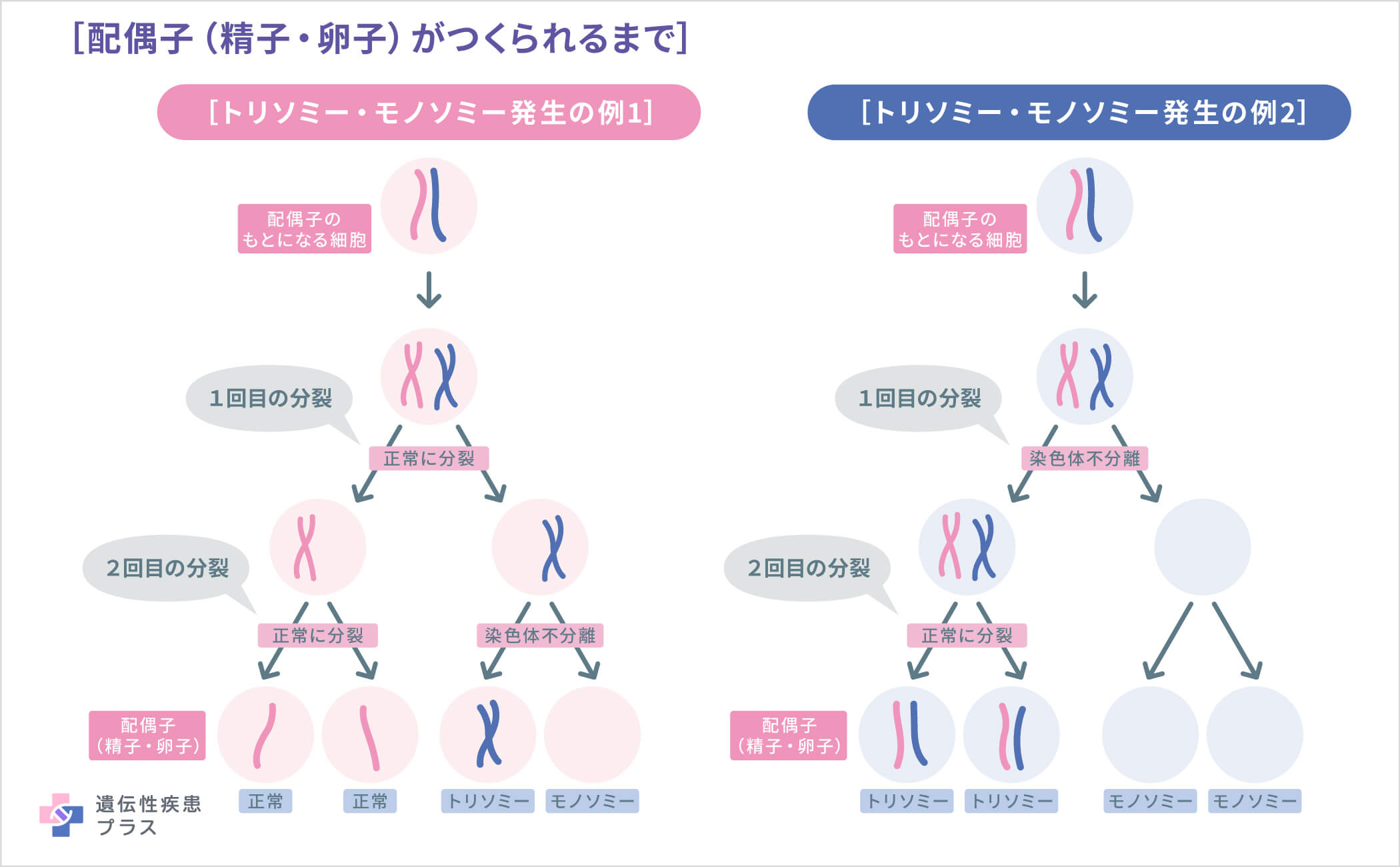
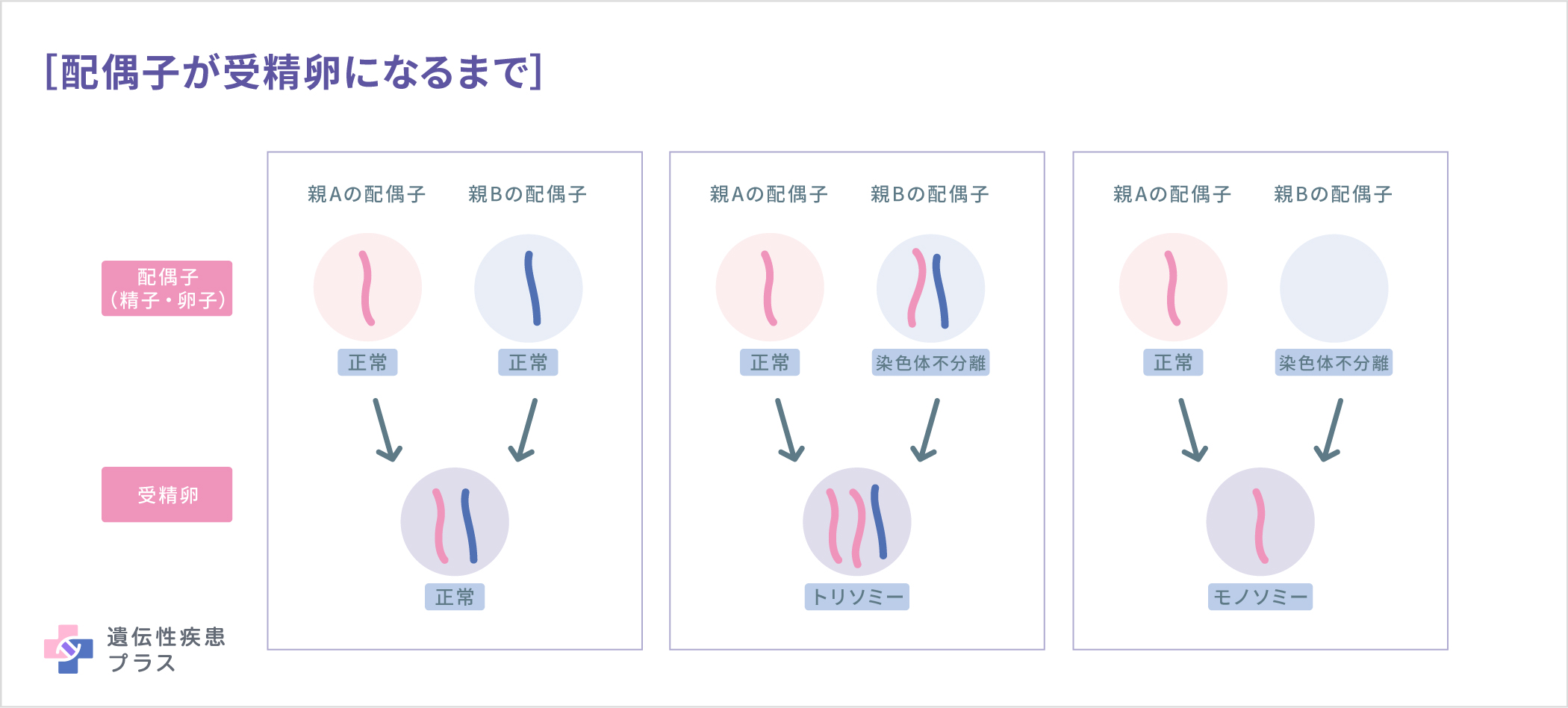
一般的にターナー症候群は、こうした卵子または精子の形成中にランダムに起こる現象が原因で発生します。そのため、ターナー症候群のほとんどの症例は遺伝しません。モザイク・ターナー症候群も遺伝しません。まれに見られる、X染色体の部分的な欠失が原因のターナー症候群は、親から子へ遺伝する可能性があります。
どのように診断されるの?
いまのところ、ターナー症候群と診断されるための、正確な定義はありませんが、低身長、思春期が遅い、不妊など、ターナー症候群が疑われる症状がある女性の場合、染色体検査が行われます。染色体検査は一般的に、採血をし、血液中に含まれるリンパ球の染色体を調べます。この検査でX染色体の全体または特定の構造異常が確認された場合、ターナー症候群と診断されます。ターナー症候群が疑われる症状が見られるものの、血液を用いた染色体検査で異常が見つからなかった場合、モザイク・ターナー症候群である可能性を考え、皮膚の細胞や性腺細胞など、別の組織を用いた染色体検査が行われる場合もあります。実際、こうした過程を経てモザイク・ターナー症候群の確定診断がついた例もあります。なお、染色体検査の精度は100%ではありません。場合によっては、FISH法、WCP解析などと呼ばれる、より詳しい遺伝子検査が行われます。
X染色体に異常が見られても、ターナー症候群が疑われる臨床症状が見られないことや、臨床症状が見られても、X染色体に異常が見つからないこともあります。こうしたケースでは、ターナー症候群とは別の診断名(X染色体長腕欠失による二次性無月経、SHOX異常症など)が付けられる場合もあります。
どのような治療が行われるの?
今のところターナー症候群を根本的に、つまり染色体の異常を治す治療はありません。現在行われている治療は、それぞれの症状に対する対症療法が中心となります。
ターナー症候群における低身長に対しては、成長ホルモンを注射する補充療法が行われます。この治療法は、有効性が認められており、治療を受けた場合、受けなかった場合に比べて平均身長は10cm程度高くなることが知られています。
卵巣機能不全に対しては、思春期が始まる10~14歳頃から、女性ホルモンの補充療法が開始され、その後継続していきます。成長ホルモンによる治療で身長が140cmに達した時点が、女性ホルモンによる治療開始の目安とされています。一般的に、「エストロゲン」「プロゲステロン」という2種類の女性ホルモンを、年齢に応じて段階的に、成人量にまで増量していきます。こうした治療により、月に1回定期的に月経が来るようにします。なお、不妊に対しては、まだ有効な治療法はありませんが、卵巣移植も試みられています。
そのほかの合併症に対しても、必要に応じてそれぞれ治療が行われます。性腺腫瘍のリスクが高いと認められた場合には、思春期前に性腺の摘出手術を受ける場合もあります。外性器の形態異常に対し、手術が行われる場合もあります。免疫系や心臓、腎臓などの合併症に対しても、適切な治療を受けていきます。知的障害がある場合には、早期から適切なサポートを受けていきます。
ターナー症候群には、患者・家族会があります。そのような場へ参加することで、病院を受診するだけでは得られない、生活に関する情報などを知る機会にもなり得ます。
どこで検査や治療が受けられるの?
日本でターナー症候群の診療を行っていることを公開している、主な施設は以下です。
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
ターナー症候群の患者会で、ホームページを公開しているところは、以下です。
参考サイト
- 難病情報センター
- 小児慢性特定疾病情報センター ターナー(Turner)症候群
- 日本内分泌学会 ターナー症候群
- MedlinePlus
- Genetic and Rare Diseases Information Center






