



どのような病気?
16p13.11微細欠失症候群は、16番染色体の一部に起こる小さな欠失を原因とする非常にまれな遺伝性疾患です。この病気は、これまでに報告された患者さんの人数が少なく、症状や特徴などまだ不明な点が多い病気です。
主な症状として、知的障害を含む全般的な発達遅延、特徴的な顔立ちのほか、てんかん、低身長、行動障害などが見られます。多くの人で言語発達の遅れや言語障害が見られるほか、多小脳回(小脳の形態異常)、ウィルムス腫瘍なども報告されています。
顔立ちや見た目の特徴として、頭部が小さい(小頭症)、下がった目尻(眼瞼裂斜下・がんけんれつしゃか)、短い鼻、低い位置にある耳、広い口、薄い上唇などが見られます。
また、英国に本部を置く希少染色体疾患サポートグループ「UNIQUE」のwebサイトに掲載の疾患ガイドには、16p13.11微細欠失症候群について、同じ欠失を持っていても症状や重症度に個人差があり、同じ家族であっても症状がさまざまであると記載されています。
16p13.11微細欠失症候群で見られる症状 |
|---|
高頻度に見られる症状 言語発達の遅れ、限局性学習障害(知的発達に遅れはないが、読み・書き・計算など特定の技能が困難)、重度の知的障害、全般的発達遅延 |
良く見られる症状 小頭症、上向きの鼻(鼻孔)、短い鼻、低身長、全般起始発作(大脳の両側からの電気信号の過剰な興奮で起こるてんかん発作) |
しばしば見られる症状 単眼症、下向きの眼瞼裂、耳介低位(低い位置の耳)、耳の前の皮膚の小さな突起物、低い鼻梁(びりょう、鼻筋のこと)、平らな人中(鼻と口の間)、口蓋裂、口唇裂、上唇が薄い、上唇の中央の曲線(キューピッドの弓とも言われる部分)が目立つ、大きな口、漏斗胸、屈指症(指をまっすぐに伸ばせない)、内反足(足が内向きや上向きに曲がる)、中足骨(足の甲にある細長い骨)の外反、神経細胞移動の異常(大脳皮質の形成異常を引き起こす)、脳梁欠損症、全前脳胞症(胎生期の前脳が正しく分割されないことによる脳の構造異常)、脳室拡大、脳波異常、感音性難聴、外耳道閉鎖症(耳の穴が閉鎖もしくは非常に狭い)、心房中隔欠損症、心室中隔欠損症、胃食道逆流症、停留精巣、筋緊張の亢進、摂食障害、強迫行動、統合失調症、自傷行為 |
16p13.11微細欠失症候群の発症頻度や日本における患者数は、はっきりとわかっていません。欧州の希少疾患情報サイト「orphanet」では、この疾患の発症頻度は100万人に1人以下で、(2011年のページ更新時点において)文献などで報告された患者さんの数は15人以下と記載されています。
また、「UNIQUE」の疾患ガイドでは、2018年のレポート作成時において16p13.11微細欠失症候群をもつ13人の会員が所属しており、医学文献にはさらに52人の患者さんが報告されていると伝えています。
一方、2021年から始まったマイクロアレイ染色体検査(染色体の検査)に含まれる微細構造異常症候群(32 疾患)の、国内での実情を把握する目的で行われた研究によれば、研究班メンバーの所属施設において診断された患者数から割り出された16p13.11(反復性)微細欠失および16p13.11重複症候群の発症頻度は3,000~7万人に1人と推定され、そこから日本における患者数は2疾患を合わせて推定数千人程度と報告されており、実際にはまだ診断されていない患者さんが多く存在する可能性も示されています。
何の遺伝子が原因となるの?
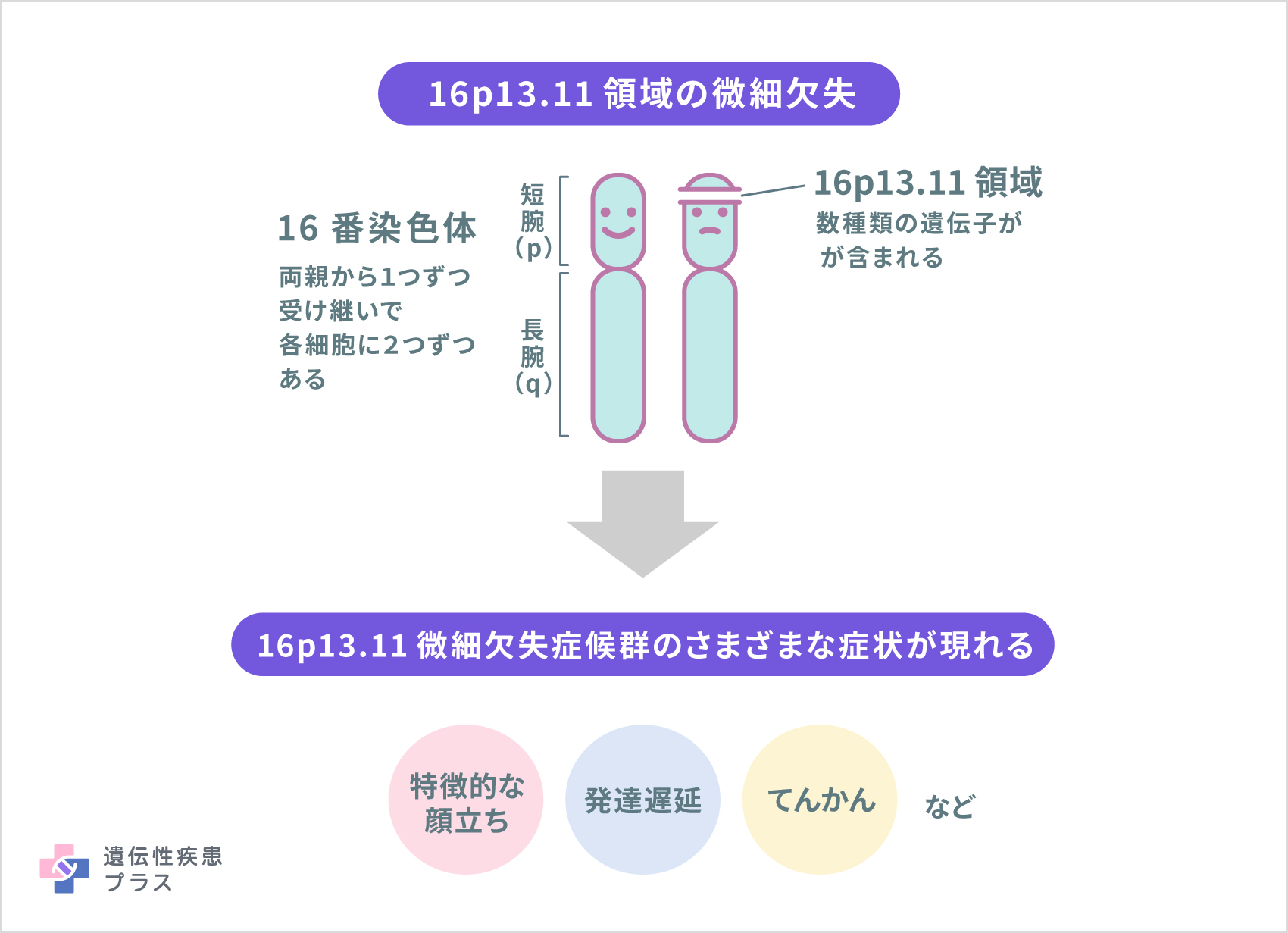
全ての細胞に存在する染色体は、1番から22番までの番号が付いた常染色体とXとYの性染色体があります。それぞれの染色体にはpで示される短腕、qで示される長腕があります。
16p13.11微細欠失症候群は、このうち16番染色体上の短腕、16p13.11と呼ばれる領域において約1.0Mbの長さで欠失(一部の遺伝子とともに消失)することが原因となり引き起こされます。欠失した部分に存在していたいくつかの遺伝子の機能が失われることがこの病気の症状の原因になると考えられます。
この領域にはNDE1、MYH11、ABCC1、ABCC6などの遺伝子が含まれています。このうち、NDE1(核分布遺伝子Eホモログ1)とNTAN1(N末端アスパラギンアミダーゼ)が神経認知などの症状と関連している可能性がありますが、詳細なメカニズムはわかっていません。
16p13.11微細欠失症候群では、両親はこの病気を発症しておらず孤発例として発症する場合と、親からこの病気の原因となる染色体の欠失を受け継ぎ、常染色体優性(顕性)遺伝形式で発症する場合があります。
常染色体優性(顕性)遺伝形式は、両親のうちのどちらかがこの病気である場合、50%の確率で発症する遺伝形式ですが、この病気では実際、軽度の症状が見られる親もしくはまったく症状がない親からこの病気の微細欠失が受け継がれ、発症する例が報告されています。そのため16p13.11微小欠失の浸透率は100%ではなく、症状が現れる表現度はさまざまであることが示唆されます(参考:不完全浸透と多様な表現度とは?)。

どのように診断されるの?
16p13.11微細欠失症候群における診断基準はまだ確立されていませんが、厚生労働科学研究難治性疾患政策研究事業「マイクロアレイ染色体検査で明らかになる染色体微細構造異常症候群を示す小児から成人の診断・診療体制の構築」研究班により、16p13.11微細欠失および重複症候群の診断基準案が作成されています。
それによれば、16p13.11微細欠失症候群の診断は、大症状として知的能力障害(IQ70未満)を認め、かつ遺伝学的検査で染色体16p13.11領域の欠失を認めた場合に、診断確定となります。症状を満たさないものの遺伝学的検査の結果が合致した場合にはこの病気の疑いありとされます。
どのような治療が行われるの?
16p13.11微細欠失症候群について、本質的な治療はまだ確立されておらず、症状に応じた対症療法が主な治療となります。
この病気は多くの症状や合併症が見られることから、それぞれの症状や問題に対応した治療が必要となります。療育やリハビリが行われる場合もあります。無症状な場合も含め、遺伝学的診断に基づく遺伝カウンセリングが大切となります。
どこで検査や治療が受けられるの?
希少遺伝性疾患の診療を広く行っていることを公開しているところは、以下です。
- 北海道立子ども総合医療・療育センター 遺伝診療科
- 東京科学大学病院 稀少疾患先端医療センター
- 慶應義塾大学病院 臨床遺伝学センター
- 東京都立小児総合医療センター
- 国立成育医療研究センター 遺伝診療科
- 昭和大学横浜市北部病院 臨床遺伝・ゲノム医療センター
※日本で16p13.11微細欠失症候群の診療を行っている医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
難病の患者さん・ご家族、支えるさまざまな立場の方々とのネットワークづくりを行っている団体は、以下です。
参考サイト
- orphanet
- Genetic and Rare Diseases Information Center
- NIH, MedGen “16p13.11 microdeletion syndrome”
- 今泉太一、山本俊至、特集・第65回日本小児神経学会学術集会シンポジウム1:マイクロアレイ染色体検査で明らかになる染色体微細構造異常症候群の診療体制を考える「研究班による実態調査結果」、脳と発達
- Rare Chromosome Disorder Support Group -Unique, 「16p13.11 microdeletions」
- 厚生労働科学研究難治性疾患政策研究事業、マイクロアレイ染色体検査で明らかになる染色体微細構造異常症候群を示す小児から成人の診断・診療体制の構築






