



どのような病気?
18q欠失症候群は、全般的発達遅滞、特徴的な顔立ち、骨格の異常などを特徴とする遺伝性疾患です。
多くの場合、胎児期もしくは生まれた時から体重が軽く成長が遅れ、低身長となります。赤ちゃんは筋肉の緊張が弱く(筋緊張低下)、てんかんの発作がみられることもあります。発達では、座る、歩く、話すなどの習得が遅れるほか、軽度から重度の知的障害が見られ、特に言葉の発達(語彙力や発語)が遅れる場合があります。また、行動上の問題として、多動性、不安、イライラなどの気分障害、うつ病、自閉症スペクトラム様の症状が見られることもあります。また、この病気の患者さんの4分の1で先天性の心疾患が見られます。
顔立ちや見た目の特徴としては、小頭症、顔の真ん中(正中)の低形成、目が奥まっている、耳が大きい、唇が厚いなどがあります。骨格の異常もあり、脊柱側弯症や後湾症のほか、内反足、外反股、第5指(小指)の湾曲なども見られます。
その他には、目の異常(斜視、近視、網膜の異常など)、難聴、甲状腺機能の低下、男性の尿道下裂(尿道の開口部が陰茎の先ではなく下側にある)などが見られることもあります。
この病気では、神経細胞の軸索と呼ばれる長い突起に巻き付く髄鞘(ずいしょう、ミエリン鞘)とよばれる構造が正常に形成されず、先天性大脳白質形成不全症と呼ばれる、大脳の白質に異常が現れる症状が見られることがあります。髄鞘は、情報の迅速な伝達を助ける役割があり、神経系の症状や発達・学習の問題が引き起こされる原因の一つとも考えられています。
この病気の予後については、合併症が安定して管理されている場合には比較的良好であるという情報もあります。
| 18q欠失症候群で見られる症状 |
|---|
良く見られる症状 顔の形の異常、低い鼻梁(びりょう、鼻筋のこと)、大耳症、下向きの口角、下顎前突、手のひらの皮膚紋理(ひふもんり、指紋のような皮膚の隆起のこと)の異常、クモ指症(指が異常に長く細い)、先細り指、小さい陰茎、内反足、扁平足、低身長、骨格の発達の遅れ、脊柱後湾症、びまん性白質の異常、髄鞘(ずいしょう)形成の異常、網膜の異常、動脈管開存症、両側の停留精巣、新生児期の筋緊張低下、協調運動不全、全般的発達遅滞、成長の遅れ、知的障害、二次成長ホルモンの欠乏 |
しばしば見られる症状 小頭症、低い前頭毛髪線、眼瞼裂斜下、内眼角贅皮(ないがんかくぜいひ、目頭部分を上まぶたが覆う)、高く目立つ鼻、団子鼻、短い人中、高口蓋、大きな口、開いた口、厚い唇、漏斗胸、細身の体形、関節の過剰可動性、両側頭頂部の狭窄、小脳形成不全、水頭症、後鼻孔狭窄、肺動脈弁の欠損または形成異常、右下行大動脈と右動脈管を伴う左大動脈弓、僧帽弁閉鎖不全、二次孔型心房中隔欠損、左右短絡、大動脈瘤、大動脈弁狭窄、心不全、甲状腺機能低下、舞踏病アテトーゼ、てんかん、感音性難聴、斜視、発育不良 |
まれに見られる症状 星細胞腫、環軸椎の異常 |
18q欠失症候群は、出生4万人に1人と推定され、これまでに300人以上の報告があります。男女比は2:3の割合で女性が多いとされています。
また、18q欠失症候群は、国の指定難病対象疾病(指定難病179)、および小児慢性特定疾病の対象疾患である「先天性大脳白質形成不全症」に含まれる病気の1つです。
何の遺伝子が原因となるの?
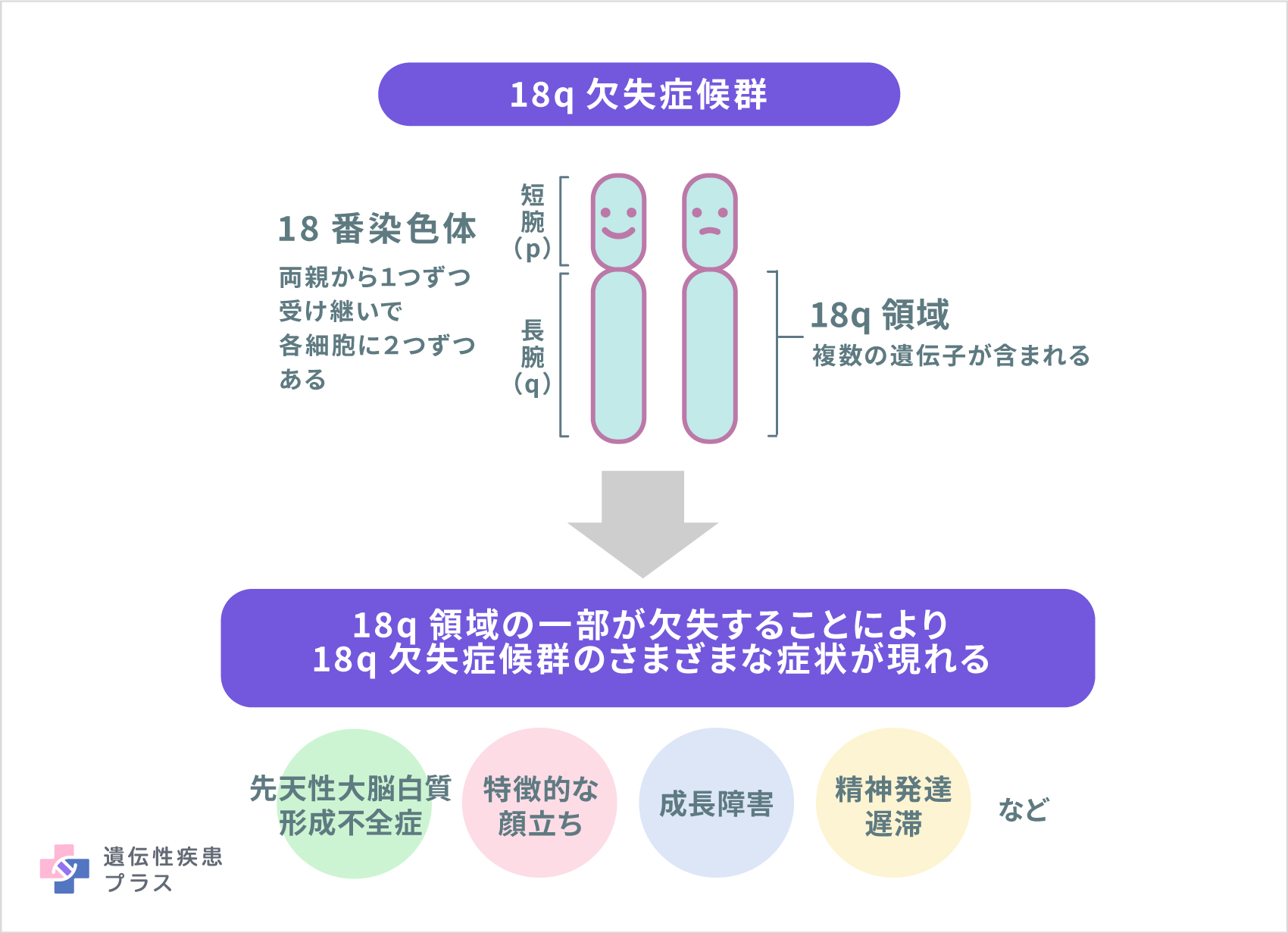
18q欠失症候群は、18番染色体の長腕(染色体の長い方の腕)の一部分が失われること(欠失)によって引き起こされます。欠失部分に存在していたいくつかの遺伝子の機能が失われることが症状の原因になると考えられます。
染色体は全ての細胞の核にあり、1番から22番までの番号が付いた常染色体とXとYの性染色体があります。それぞれの染色体にはpで示される短腕、qで示される長腕があります。
この病気において欠失する領域や大きさは患者さんによってさまざまですが、18q21と呼ばれる領域から染色体の末端の部分を欠失している人がほとんどとされます。18q23領域にあるMyelin basic protein(MBP)遺伝子の欠失は、18q欠失症候群にみられる先天性大脳白質形成不全の原因とされています。
18q欠失症候群の症状の多くはハプロ不全と呼ばれる遺伝子の変異が関係しています。ハプロ不全は、正常な遺伝子が1コピーとなった場合に機能が十分保てない遺伝子に変異が起こる場合に引き起こされます。ヒトの遺伝子は両親から受け継ぎ2コピーずつあるため、多くの遺伝子は一方に異常があっても、1コピーの正常な遺伝子が補うことができます。この病気で欠失した染色体部分に含まれる遺伝子の中に2コピーないと機能が保てない遺伝子が含まれ、それらによるハプロ不全によってそれぞれの症状が引き起こされると考えられます。しかし、欠失した遺伝子とそれぞれの症状にどのような関連があるのかについて多くはまだわかっていません。
18q欠失症候群では、ほとんどの場合において両親はこの病気を発症しておらず、患者さんは新たに染色体が欠失したことによる孤発例です。また、病気を発症していない親の染色体において均衡型転座(異なる染色体間で一部が入れ替わるが全体は重複や欠失が無い転座)があり、子どもが引き継ぐ時に不均衡型転座(重複や欠失がある転座)となることでこの病気を発症することがあります。親がこの病気を発症している場合には、18q欠失症候群は常染色体優性(顕性)遺伝形式で遺伝し、子どもが18q欠失症候群となる確率は50%です。

どのように診断されるの?
18q欠失症候群の診断基準は、1)成長障害(特に低身長)、2)発達遅滞、3)MRI検査で白質に病的な高信号が見られる、4)遺伝学的検査で18番染色体の長腕の欠失が認められること、の4つを満たした場合にこの病気であると診断されます。
どのような治療が行われるの?
18q欠失症候群の根本的な治療法はないため、それぞれの症状に応じた対症療法が行われます。また、早期に遺伝学的な検査による診断の確定が重要であり、乳幼児期から定期的な医療管理、療育的な支援、リハビリを受けることが望まれ、聴力検査も重要とされます。先天性心疾患の合併には手術などの治療が行われる場合もあります。
どこで検査や治療が受けられるの?
日本で18q欠失症候群の診療を行っていることを公開している、主な施設は以下です。
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
難病の患者さん・ご家族、支えるさまざまな立場の方々とのネットワークづくりを行っている団体は、以下です。





