



どのような病気?
22q11.2欠失症候群は、22番染色体の中央付近の22q11.2という部分に、顕微鏡では見えないくらい小さな欠失(微細欠失)があるために、さまざまな症状が起こる遺伝性疾患です。
多くみられる症状は、先天性心疾患、精神発達遅延、特徴的な顔立ち、胸腺の形成不全による免疫低下、鼻声、低カルシウム血症などです。心疾患のうち多く見られるのは「ファロー四徴症」です。また、ファロー四徴症の患者さんのうち約15%は22q11.2欠失症候群です。ファロー四徴症は、「心室中隔欠損:左右の心室を分ける心室中隔という仕切りの壁に大きな穴がある」「大動脈騎乗:全身へ血液を送る大動脈が左右の心室にまたがっている」「肺動脈狭窄・漏斗部狭窄:肺へ血液を送る肺動脈の右心室からの出口(漏斗部)が肺動脈弁と一緒に狭くなる」「右心室肥大:左右の心室の圧が等しくなり、右心室が肥大する」の4つの特徴がみられるものです。
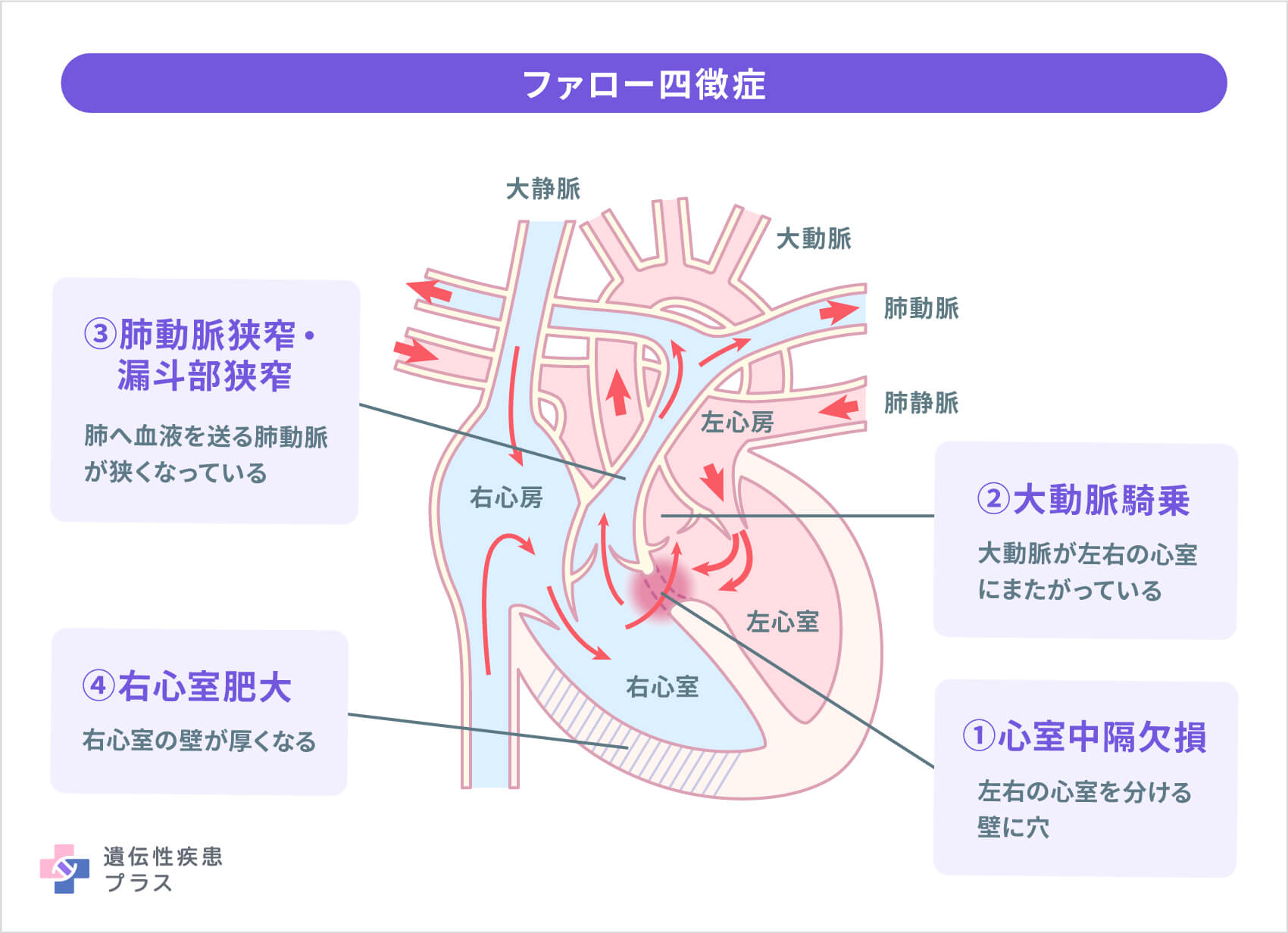
肺動脈狭窄の度合いによって、低酸素血症のために唇や爪が紫色になる「チアノーゼ」が起こる時期や程度が異なります。約3人に1人は生後1か月以内、約3人に1人は生後1か月~1年以内の乳児期に、残りの約3人に1人は1歳以後の幼児期にチアノーゼが現れます。小さいうちは、泣いた後や運動をした時に見られ、成長にともない常に見られるようになります。生後2か月以後には、急に不機嫌になり、チアノーゼと呼吸困難が強くなり、場合によっては意識を失ったり、全身のけいれんを起こしたりすることがあります。これは、「チアノーゼ発作(低酸素発作)」というもので、就寝後や午前中に見られることが多く、重くなると一日中起きるようになります。多くの場合は10分程度で自然に改善しますが、長時間続くと命に関わります。チアノーゼが見られるようになってから6か月以上経つと、手足の指先が肥大して、太鼓のばちのような形になります(ばち指)。
22q11.2欠失症候群でみられる心臓や血管の症状は、その度合いによって命に関わることもあります。多くの場合、乳児期に心臓の手術を受けますが、手術を受けずに小学校に上がった場合はチアノーゼが強く、少しの運動で息が切れるため、学校や日常生活での制限が強くなります。22q11.2欠失症候群の子どもたちの多くは、成長や言語などの発達に遅れがみられ、軽度の知的障害や学習障害のある子どももいます。このような精神発達遅延は、小学校前から目立ってきます。人によっては、コミュニケーションに影響のある発達障害も見られます。
| 22q11.2欠失症候群でみられる症状 |
|---|
99~80%でみられる症状 大動脈弓の形態異常、肺動脈弁の形態異常、咽頭の異常、心房中隔欠損症、球状の鼻、口蓋裂、伝音難聴、失語症、内眼角贅皮、胸腺の形成不全、免疫不全、耳介低位、筋緊張低下、鼻声、扁平頭蓋底、高い鼻梁、内眼角外方偏位、ファロー四徴症、総動脈幹症、上向きの眼瞼裂、心室中隔欠損症、広い鼻梁 |
79%~30%でみられる症状 扁桃腺の異常、痤瘡、細くて長い指、注意欠如・多動症、虫歯、慢性中耳炎、便秘、角膜血管新生、全般的発達遅滞、低カルシウム血症、副甲状腺機能低下症、T細胞の機能不全、軽度の知的障害、長い顔、長い人中、平坦な頬、筋肉痛、後頭脊髄髄膜瘤、過剰に折れ込んだ耳輪、後部胎生環(目の異常)、眼瞼下垂、腎形成不全、脂漏性皮膚炎、短い首、低身長、小さな耳たぶ、限局性学習障害、テタニー(手足のけいれん)、薄い上唇 |
29%~5%でみられる症状 大動脈弁の形態異常、異常分葉肺、胸部の形態異常、歯のエナメル質の異常、子宮の奇形、無神経節性巨大結腸症、鎖肛、不安、不整脈、関節炎、ぜんそく、無気肺、自閉症、自己免疫、双極性障害、便失禁、白内障、後鼻孔閉鎖症、胆石症、慢性閉塞性肺疾患、停留精巣、うつ病、下向きの眼瞼裂、成長障害、乳児期の摂食困難、手や足の多指症、胃食道逆流症、消化管出血、緑内障、水頭症、両眼隔離症、高血圧クリーゼ、甲状腺機能亢進症、皮膚低色素斑、尿道下裂、甲状腺機能低下症、鼠径ヘルニア、腸回転異常症、出生前発育不全、関節が可動域を超えて動く、喉頭軟化症、小頭症、小顎症、小眼球症、多発性頭蓋骨縫合部早期癒合症、小さな口、肥満、視神経萎縮、膝蓋骨の脱臼、動脈管開存症、多発性囊胞腎、羊水過多症、紫斑、網膜細動脈ねじれ症、統合失調症、脊柱側弯症、発作、短い人中、脾腫、斜視、内反足、血小板減少症、三尖弁閉鎖症、尖頭症、臍ヘルニア、静脈瘤、膀胱尿管逆流症 |
4%~1%でみられる症状 行動異常、くぼんだ目、アーチ型の眉、とがった顎、平坦な人中、鼻翼未発達 |
22q11.2欠失症候群は、4,000~6,000人に1人の頻度で発症すると推定されており、日本には約4,500人の患者さんがいます。22q11.2欠失症候群の赤ちゃんが生まれやすくなると言い切れるような環境や要因は、今のところ見つかっていません。
22q11.2欠失症候群の徴候や症状は非常に多様であるため、家族内に同じ病気の人がいる場合でも、症状や重症度は同じとは限りません。かつては症状ごとに、別の病気として説明されていましたが、遺伝的な原因が特定され、全て「22q11.2欠失症候群」の一部であると判明しました。それが、この病気にたくさんの別名がある理由です。今では、混乱を避けるために、遺伝的な根拠に基づき「22q11.2欠失症候群」と呼ばれるようになっています。
22q11.2欠失症候群は、国の指定難病対象疾患(指定難病203)、および、小児慢性特定疾病の対象疾患です。
何の遺伝子が原因となるの?
22q11.2欠失症候群は、ほとんどの場合、各細胞について、両親から1つずつ計2つ受け継いだ22番染色体のどちらか一方に、顕微鏡では見えない程度の小さな欠失(微細欠失)があることで起こります。微細といっても、約300万塩基対の配列が欠けており、その部分には約30~40の遺伝子が含まれています。欠失している部分がもう少し短い人もいます。
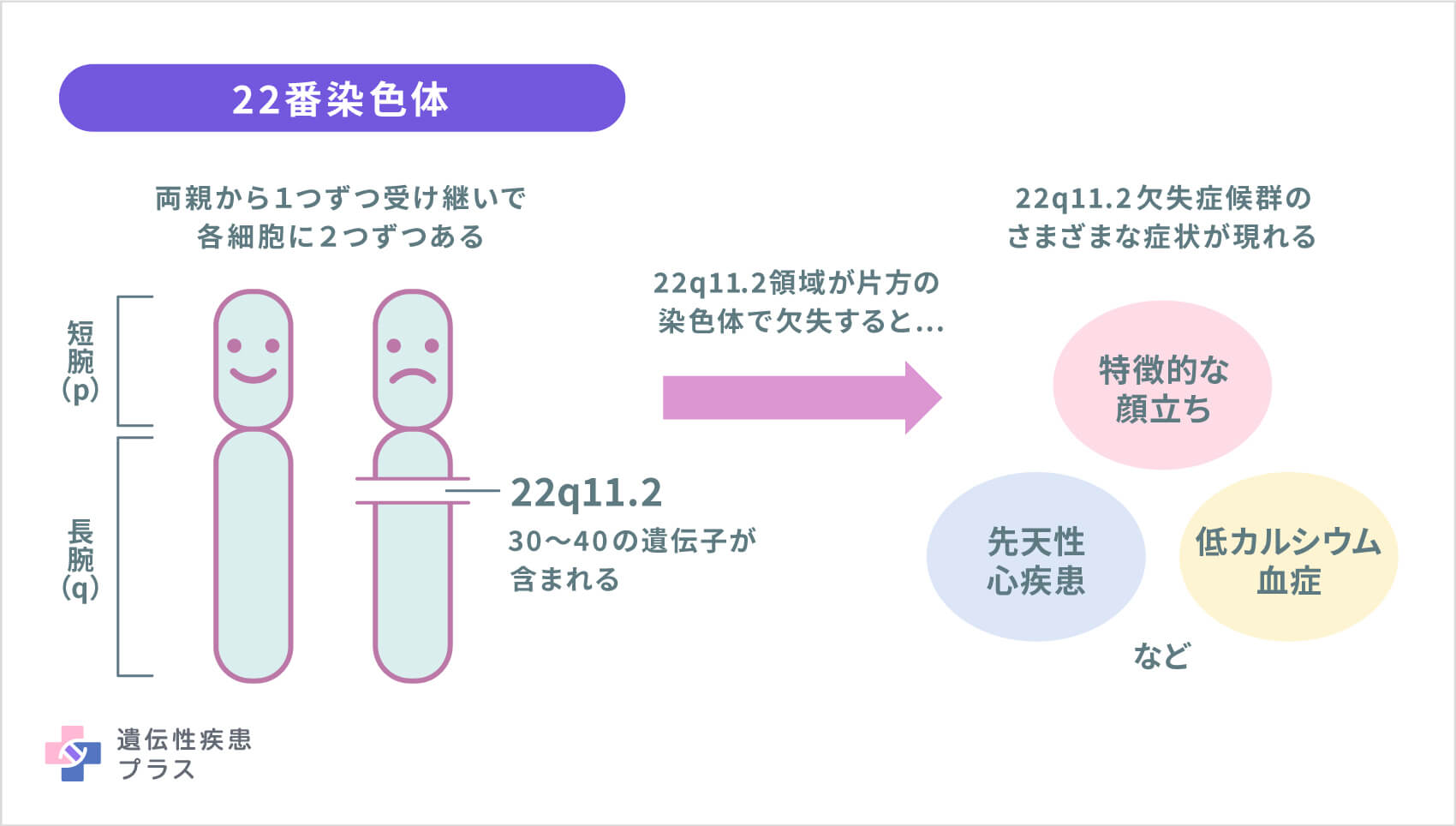
この部分にある複数の遺伝子は、まだ役割が十分にわかっていないものが多いのですが、この領域に含まれる複数の遺伝子の欠失により、22q11.2欠失症候群のさまざまな症状が現れている可能性が考えられています。
これまでの研究により、今のところ、この部分に存在するTBX1遺伝子が片方の22番染色体で欠失していると、心疾患、口唇裂、独特の顔立ち、難聴、低カルシウム血症など、22q11.2欠失症候群に特徴的な症状が起こるとわかっています。TBX1遺伝子の欠失が、22q11.2欠失症候群でみられる行動上の問題にも関連する可能性があると示した研究も、複数あります。また、同じ領域にあるCOMT遺伝子の欠失も、行動上の問題や精神疾患に関連する可能性があると示されています。
22q11.2欠失症候群は、各細胞に2つずつある22番染色体の1つに微細欠失があれば発症するため、常染色体優性(顕性)遺伝形式で親から子へ受け継がれると考えられています。この欠失がある人に子どもがいる場合、その欠失を受け継ぐ確率は50%です。ただし、親から子へ22q11.2欠失症候群が受け継がれている人は症例の約10%程度で、残りの大半は、家系に同じ病気の人がいるわけではなく、生殖細胞(卵子・精子)が作られる過程、もしくは受精後初期の発育過程で生じた変異が原因で発症した人たちです。

どのように診断されるの?
22q11.2欠失症候群には、医師が22q11.2欠失症候群と診断するための「診断基準」があります。したがって、病院へ行き、必要な問診や検査を受けた後、主治医の先生がそれらの結果を診断基準に照らし合わせ、結果的に22q11.2欠失症候群か否かの診断をすることになります。
具体的には、副甲状腺低形成による低カルシウム血症がもたらす症状、胸腺発達遅延もしくは無形成による免疫力低下(易感染性)、先天性心疾患(ファロー四徴症、円錐動脈管心奇形、大動脈弓離断、右大動脈弓、右鎖骨下動脈起始異常等の心奇形など)、特徴的な顔立ち(口蓋裂、低位耳介、小耳介、瞼裂短縮を伴う眼角隔離症、短い人中、小さな口、小顎症など)、精神発達遅滞、言語発達遅滞など、特徴的な症状が複数見られた場合に、「22q11.2欠失症候群の疑い」となります。このとき重要な検査所見は、以下の4つです。
- 低カルシウム血症、副甲状腺機能低下
- T細胞数の減少および機能低下
- B細胞数は正常、免疫グロブリン値は正常か減少
- 画像検査や心カテーテルによる心奇形の同定
22q11.2欠失症候群の疑いとなった場合、染色体検査(FISH法やaCGH法と呼ばれる方法で行われる)で、22q11.2欠失が認められると、22q11.2欠失症候群と確定診断されます。
どのような治療が行われるの?
今のところ、22q11.2欠失症候群を根本的に、つまり、遺伝子から治すような治療法は見つかっていません。一方で、症状を管理する方法はあります。新生児期から、その人ごとに適した手術計画が立てられ、症状に基づき、生活指導や治療を、生涯にわたって受け続けていきます。
治療は基本的に、心臓手術です。これは、乳児期に起こるチアノーゼ発作や運動制限を改善するために行われるものです。ファロー四徴症で高度肺動脈狭窄または肺動脈閉鎖がある場合には、新生児期に、鎖骨下動脈と肺動脈をつなぐ「ブラロックータウシッヒ短絡手術」が行われる場合があります。ファロー四徴症に対しては多くの場合、心室中隔欠損を閉鎖して狭い右室流出路を拡大形成する「心内修復術」が、生後6か月~2歳の間に行われます。心内修復術には自己弁温存法、右室流出路パッチ拡大術、ラステリ手術の3通りがあり、個人に適した手術が選択されます。そのほか、状態に応じて、例外的な手術が行われる場合もあります。
小児期に心内修復術が行われた後、血液の流路が再び狭くなった場合には、バルーンカテーテルやステントで拡大する「カテーテル治療」が行われる場合があります。また、大動脈から肺動脈に異常なバイパス血管ができた場合、コイル塞栓術が行われることがあります。退院後数年してから、右心不全を生じ、それに対する手術が行われる場合もあります。ラステリ手術は、人工血管などの導管を使って右室から肺動脈へ通路を作成する方法ですが、成人期にラステリ手術後の導管に狭いところができて、導管を交換する手術が必要になることもあります。
心内修復術を受けた後は、術後の状態にもよりますが、普通に日常生活を送れるようになります。ただし、激しい競技スポーツや運動は制限されます。運動をする場合は、準備運動や整理運動をしっかり行い、運動中の水分補給も忘れないようにすることが大切です。
心臓手術以外には、低カルシウム血症に対する治療、免疫不全状態に応じた感染症予防と治療などが行われます。また、精神発達遅延がある患者さんでは、例えば学校生活や就職に際する職業訓練など、学校や行政機関と相談して社会支援を受けていく場合があります。
妊娠・出産は、術後の合併症が重くなければ可能です。お子さんを望まれる場合には、主治医と事前によく相談して、心機能の評価をしていくことが大切になります。お子さんへの病気の遺伝に関して心配なことなどがあれば、主治医や遺伝カウンセラーに相談しましょう。
どこで検査や治療が受けられるの?
日本で22q11.2欠失症候群の診療を行っていることを公開している、主な施設は以下です。
- 東京大学医学部附属病院
- 国立成育医療研究センター 遺伝診療科
- 東京都立小児総合医療センター
- 慶應義塾大学病院 精神・神経科
- 東京女子医科大学病院 ゲノム診療科
- 東京科学大学病院 遺伝子診療科
- 昭和大学横浜市北部病院 臨床遺伝・ゲノム医療センター
- 大阪母子医療センター 遺伝診療科
- 兵庫県立こども病院
- 岡山大学病院 口唇裂・口蓋裂総合治療センター
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
難病の患者さん・ご家族、支えるさまざまな立場の方々とのネットワークづくりを行っている団体は、以下です。
参考サイト
- 難病情報センター
- 小児慢性特定疾病情報センター 胸腺低形成(ディ・ジョージ(DiGeorge)症候群/22q11.2欠失症候群)
- MedlinePlus
- Genetic and Rare Diseases Information Center






