



どのような病気?
アンジェルマン症候群は、重度の発達の遅れ、言葉が出ない、てんかん、容易によく笑う、動き方がぎこちないなど、主に神経系に関連した症状が見られる遺伝性疾患です。
発達の遅れは、乳児期の後半から気づかれることが多く、一人歩きができるようになるのは平均すると5歳くらいとされます。精神発達や発話については、意味のある言葉を話すようになることは大人になってもほとんどありませんが、理解の発達は比較的良いとされています。てんかんの発作は、およそ8割に見られ、乳児期には熱性けいれん、その後はさまざまなタイプのてんかん発作が起こることが知られます。薬を服用しても発作が治まりにくい難治性てんかんがあると、予後に大きく影響します。
行動の特徴としては、ちょっとしたことで容易によく笑う、落ち着きがない、旺盛な好奇心、水やビニールなどキラキラしたものに対する興味などがあります。乳児期から幼児期には睡眠障害の合併が多く、夜間中途覚醒、入眠の遅れ、早期覚醒などがみられますが、行動や睡眠障害は年齢とともに改善していく傾向があります。内臓に関する合併症はほとんどありません。また、大人になるとあまり動かなくなり、肥満になりやすくなることが知られています。その他の症状としては、低色素症、特徴的な顔立ち(尖った顎、大きな口)などがあります。
| アンジェルマン症候群で見られる症状 |
|---|
高頻度に見られる症状 重度の全般的発達遅滞、重度の知的障害、言語発達遅滞、神経学的発語障害、発話困難、てんかん発作、不適切な笑い、行動障害、自閉症的行動、自傷行為、活動過剰、運動発達遅滞、歩行時の広い歩隔(左右の足の間の幅)、運動失調、睡眠障害、小頭症、振戦(意図しない震え)、皮質性小脳萎縮症 |
良く見られる症状 睡眠-覚醒サイクル障害、手を上下に羽ばたかせるような動き、多食症、肥満、金髪、低色素の虹彩、低色素の皮膚、特徴的な顔立ち、斜視、乱視、大きい口、舌突出、よだれ(唾液分泌過多)、脊椎側弯症、乳児期の筋緊張低下、胃食道逆流、摂食障害、便秘、消化管の異常 |
しばしば見られる症状 発語欠損、思春期早発症(女児)、遅い初潮、てんかん重積状態、ミオクローヌス(間代性筋けいれん)、全身性ミオクロニー発作、強直間代発作(けいれんを伴うてんかん発作)、非定型欠神発作(てんかん発作の一種)、脱力発作(てんかん発作の一種)、他人と目を合わせない、不安症、状況によらず幸せそうな態度、攻撃的行動、歩行不能、外反足、後頭部扁平、眼瞼下垂、円錐角膜、視神経乳頭蒼白、視神経委縮、遠視、近視、弱視、眼振、隙間の多い歯、舌突出、下顎突出、嘔吐、哺乳障害、乳児期の胃瘻(いろう)造設術治療、経鼻管栄養治療、嚥下障害、大脳髄鞘(ずいしょう)異常 |
非常にまれに見られる症状 |
アンジェルマン症候群は出生1万5,000人に1人程度の頻度であると推定され、難病情報センターによると日本で500~1,000人程度の患者さんが確認されています。また、小児慢性特定疾病情報センターによると、日本には3,000人以上の患者さんがいると考えられています。アンジェルマン症候群は指定難病(指定難病201)および小児慢性特定疾病となっています。
何の遺伝子が原因となるの?
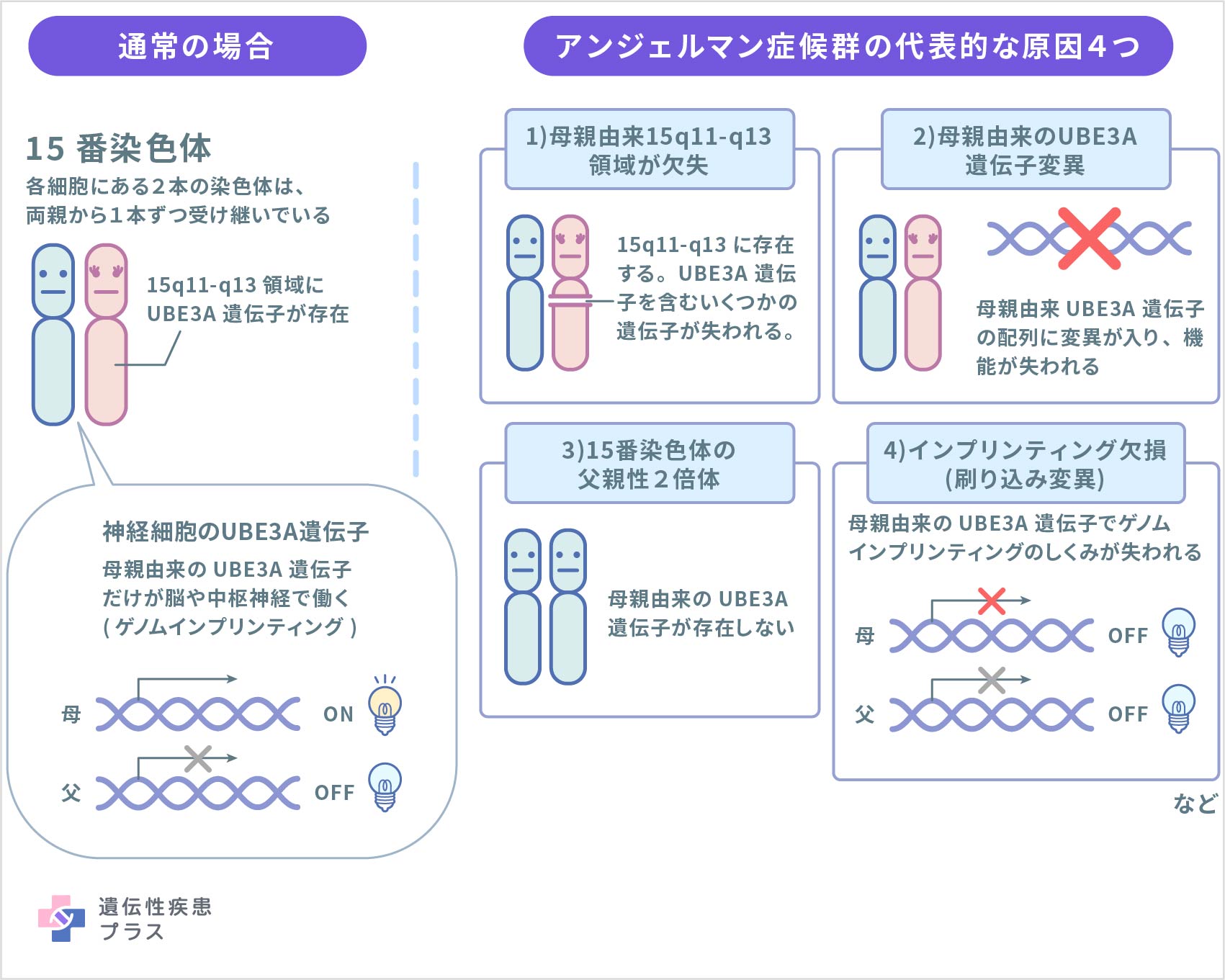
アンジェルマン症候群は、15番染色体15q11.2領域に位置するUBE3A遺伝子の機能喪失が原因で発症すると考えられています。UBE3A遺伝子は、ユビキチンリガーゼE3A(UBE3A)と呼ばれるタンパク質の設計図となる遺伝子です。ユビキチンリガーゼは、細胞内で不要になり壊されるべきタンパク質が、正しく分解されるために必要な酵素として働きます。その中でも、UBE3Aは神経系の正常な発達と機能に重要な役割を果たしていると考えられています。ヒトの記憶や学習は、シナプス可塑性と呼ばれる、経験に応じて神経細胞同士のつながりやつながる強さが変化することが基盤となっていますが、このシナプス可塑性に重要なタンパク質恒常性(タンパク質合成や分解が正しく行われる)の制御を、UBE3Aが担っていると考えられています。
ヒトは通常、父親と母親から染色体を1本ずつ受け継ぎ、遺伝子は父母由来の2本が1セットで存在しています。遺伝子の多くは、どちらの親から受け継がれた遺伝子も同じように働きますが、ある特定の遺伝子は、ゲノムインプリンティングと呼ばれる仕組みによって父親由来の遺伝子だけ、もしくは、母親由来の遺伝子だけが機能するように決められています。UBE3A遺伝子は、このゲノムインプリンティングの仕組みにより、体のほとんどの組織では父母由来の両方の遺伝子が働きますが、脳や中枢神経系の神経細胞においては、母親から受け継いだ遺伝子だけが働くように決められています。そのため、何らかの原因により母親由来のUBE3A遺伝子の機能が失われた場合、脳内では機能できるUBE3Aが存在せず、それがこの病気を引き起こすと考えられています。このUBE3A遺伝子の喪失原因として、以下の4つが報告されています。
1)母親由来の15番染色体の微細欠失
欠失とは染色体や遺伝子の一部が消失することです。母親由来の15番染色体の15q11-q13領域において、顕微鏡では確認できないレベルのUBE3A遺伝子を含む小さい欠失が起こることが病気の原因となります。アンジェルマン症候群のおよそ70%がこの原因によるとされています。
2)母親由来のUBE3A遺伝子変異
母親由来のUBE3A遺伝子の変異もアンジェルマン症候群の原因となり、全体の10%程度を占めると考えられています。
3)15番染色体における父親性2倍体
染色体は通常、父親と母親から1本ずつ受け継ぎますが、まれに染色体が父親、もしくは母親のみから2本とも受け継がれる場合があり、片親性2倍体もしくは片親性ダイソミーと呼ばれます。父親から2本とも受け継ぐ場合を父親性2倍体と呼び、15番染色体において父親性2倍体が起こると、この染色体に含まれるUBE3A遺伝子は2つとも父親由来となり、母親由来の遺伝子が存在しないことが病気の原因となります。父親性2倍体はアンジェルマン症候群の原因の5%程度を占めると考えられています。
4)インプリンティング欠損(刷り込み変異)
ゲノムインプリンティングによる制御に変異が生じ、本来の親由来の働きと反対の働きとなる変異です。母親由来で機能するインプリンティング遺伝子であるUBE3Aが、父親から由来したような状態に変異する結果、母親由来のUBE3A遺伝子が機能しなくなることで病気が発症します。全体の5%程度を占めると考えられています。
一方で、アンジェルマン症候群のおよそ10%において、遺伝的な原因はまだはっきりとわかっておらず、他の遺伝子や染色体が関与する可能性も考えられています。
また、15番染色体の15q11-q13領域のUBE3A遺伝子付近に存在するOCA2遺伝子機能の喪失は、この病気の特徴である明るい髪の色や色白の肌と関連があると考えられています。
アンジェルマン症候群の多くは卵子と精子が形成されるときにランダムに起こる現象が原因で発生するため、親からの遺伝ではなく孤発例であるとされます。病気の原因が母親由来の15番染色体の微細欠失、もしくは父親性2倍体である場合には親からの遺伝でこの病気になることはほとんどありません。原因が母親由来のUBE3A遺伝子変異もしくはインプリンティング欠損である場合には、子どもに遺伝する場合があり、遺伝カウンセリングを受けることが推奨されます。
アンジェルマン症候群と原因遺伝子領域が関連している病気として、プラダー・ウィリ症候群があります。プラダー・ウィリ症候群は、15番染色体15q11-q13領域での父親由来の遺伝子の機能喪失が原因によって引き起こされます。
どのように診断されるの?
アンジェルマン症候群の主な症状(主要臨床症状)として、1)容易に引き起こされる笑い(ちょっとしたことでよく笑うなど)、2)失調性歩行(ぎこちない動きなど)、3)下顎突出を含む特徴的な顔立ち、4)精神発達遅滞(言葉が出ないなども含む)、5)てんかん発作という5つの症状があります。
この病気の診断は、遺伝学的検査によって、15番染色体の15q11.2-15q11.3領域に、欠失、片親性二倍体、もしくはUBE3A遺伝子の変異や刷り込み変異が確認され、上記の症状の3)と4)が両方みられる場合に診断確定となります。
一方で、アンジェルマン症候群の一部では遺伝子の変異がはっきり見つからない場合もあります。このため遺伝子の変異が認められない場合でも主要臨床症状があれば臨床的な所見を基に診断されることもあります。
どのような治療が行われるの?
アンジェルマン症候群については、本質的な治療はまだ確立されておらず、てんかんや睡眠障害など、症状に応じた対症療法が主な治療となります。
てんかんに関しては抗てんかん薬、睡眠障害には睡眠薬などの投薬治療、発達の遅れに対しては、理学療法、言語療法など、療育的な支援が行われます。
生活全般について介護が必要となるので療育(発達支援)を活用するなど、福祉的なサポートの利用が勧められます。
どこで検査や治療が受けられるの?
日本でアンジェルマン症候群の診療を行っていることを公開している、主な施設は以下です。
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
アンジェルマン症候群の患者会で、ホームページを公開しているところは、以下です。






