



どのような病気?
自己貪食空胞性ミオパチーは、体の多くの筋肉の筋線維内において、自己貪食空胞(じこどんしょくくうほう)と呼ばれる特徴的な細胞内の小胞が見られる遺伝性疾患で、主な症状としては、骨格筋障害、心筋障害、知的障害などがあります。この病気の中には「ダノン病」「過剰自己貪食を伴うミオパチー(XMEA)」などが含まれ、病型によって見られる症状や重症度に違いがあります。
ダノン病は、進行性の筋力低下・筋萎縮のほかに重篤な心筋症が見られ、予後が悪いとされている疾患です。当初、ダノン病は症状や特徴が似ていることから糖原病II型であるポンペ病の変種として分類されましたが、その後この病気は解糖系の酵素を原因とするものではないことがわかり、糖原病とは別の病気とされています。
この病気は、性別によって症状や重症度に違いがあり、男性の方が重症となることが多くなります。発症年齢も男性の方が早く、ほとんどの場合男性では小児期~青年期、女性は成人期の初期に症状が見られ始めます。
よく見られる症状の中で、特に命に関わるものは、進行性の重症な心筋症(肥大型心筋症、拡張型心筋症)や不整脈です。肥大型心筋症は、心臓の筋肉(心筋)が分厚く肥大して心臓の動きが悪くなり、拡張型心筋症は反対に、心筋が薄くなって広がって大きく(拡張)なることで心臓の動きが悪くなります。男性では肥大型心筋症が見られる場合が多く、心臓移植が必要となる場合もあります。まれに、症状が進行するにつれて拡張型心筋症が発症する場合があります。女性は男性よりも心筋症を発症する割合は低いですが、心筋症がある場合には、肥大型心筋症は男性より少なく、拡張型心筋症の発症は男性より高いとされます。また、動悸、不整脈、胸痛などの心臓に関連した症状も見られ、男性の場合はほとんどにおいて心臓伝導障害(心拍を制御するための電気信号の異常)が生じます。これらの症状が進行することで、心不全となり若い時期に命を落とすことも多い病気です。
骨格筋の障害は、ダノン病ではほとんどの男性と半数の女性に見られます。通常は腕、肩、首、太ももなどで筋力低下が見られ、歩行障害や運動発達の遅れにつながる可能性があります。男性では、筋肉に障害があると高くなる、クレアチニンキナーゼという検査値の値が高くなります。
その他の症状としては、多くの男性では軽度の学習障害や知的障害が見られますが、女性ではほとんど見られないこともあるなど、その程度は個人によりさまざまです。また、網膜症を発症し、その後視覚障害を引き起こす場合もあります。
ダノン病でよく見られる症状(男女別の割合)
症状 | 男性で見られる割合 | 女性で見られる割合 | 補足 |
|---|---|---|---|
肥大型心筋症 | 96% | 30~70% | 女性は、肥大型心筋症となる可能性が男性よりも低い |
拡張型心筋症 | 4% | 30~50% | 男性では肥大型心筋症が進行した結果拡張型心筋症を発症することがある |
心臓伝導障害 | >80% | 60~100% |
|
骨格筋障害 | 80~90% | 12~50% | 通常、女性は進行性ではない |
知的障害 | ~80% | ~10% | 知的障害の程度は通常軽度で、特に女性ではさまざま |
網膜症 | ~20% | ~20% | 詳細な眼科検査が必ず行われている訳ではないため実際の割合はさらに高い可能性がある |
過剰自己貪食を伴うミオパチー(XMEA)は、進行性の筋力低下・筋萎縮が認められる疾患です。筋力低下により、日常の生活や動作に影響を及ぼすことがありますが、心筋症や他の症状は見られません。また、症状が見られるのはほとんどの場合男性のみで女性は無症状とされます。
この病気では多くの場合5~10歳くらいの小児期に症状が見られ始め、次第に階段の上り下りや走ることなどが困難になるなどの症状が見られ始めます。症状が大きくみられる筋肉に関しては、肩、腰、太ももなど体の中心に近い部分から筋力低下が始まることが多いとされ、関節の拘縮などが見られることもあります。60歳以上になると車いすが必要になる人も多くなります。ダノン病と比較すると症状は軽度で進行も遅く、通常、命に関わるような症状は見られません。
これ以外の自己貪食空胞性ミオパチーに含まれる疾患として、乳児型自己貪食空胞性ミオパチー、成人発症型の(多臓器障害を伴う)自己貪食空胞性ミオパチーなどがこれまでに報告されていますが、いずれも非常にまれな疾患で、詳細はあまりわかっていません。
自己貪食空胞性ミオパチー全体の正確な発症頻度や日本国内における患者数はわかっていません。ダノン病はこれまでに海外を含めておよそ100家系以上の報告があり、国内でも約30家系確認されています。過剰自己貪食を伴うミオパチー(XMEA)は、世界でも約15家系ほどの報告のみで、欧米からの報告がほとんどとされています。
自己貪食空胞性ミオパチーは、国の指定難病対象疾病(指定難病32)となっています。
何の遺伝子が原因となるの?
自己貪食空胞性ミオパチーは、含まれるそれぞれの疾患ごとに原因となる遺伝子が異なります。
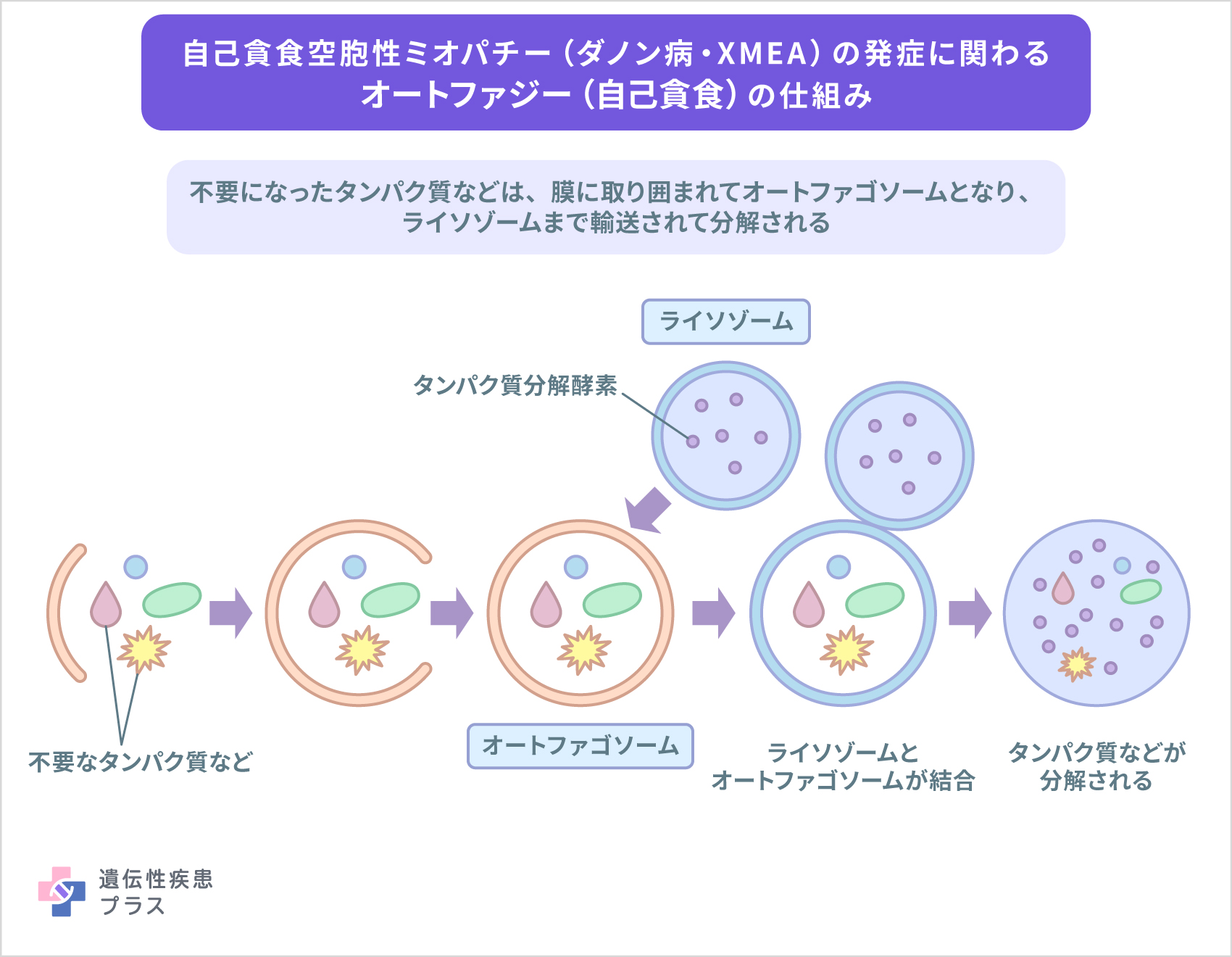
ダノン病は、X染色体のXq24領域に存在するLAMP2遺伝子の変異によって引き起こされます。LAMP2遺伝子は、ライソゾーム膜タンパク質2(LAMP2タンパク質)と呼ばれる、細胞小器官であるライソゾームの膜に存在するタンパク質の設計図となります。ライソゾームは、細胞の内外から取り込まれた物質を分解しリサイクルする機能を持ちます。細胞内で、リサイクルする物質をライソゾームまで輸送するのは、オートファジー(自己貪食)と呼ばれる仕組みで、オートファジーではオートファゴソームと呼ばれる液胞のような細胞構造が、特定の物質を包み込んでライソゾームまで輸送します。ライソゾームとオートファゴソームの膜同士が融合することで分解が行われます。
LAMP2タンパク質の詳細な役割はわかっていませんが、これまでの研究から、このライソゾームとオートファゴソームの融合に関与しているのではないかと考えられています。
LAMP2遺伝子が変異することによって、LAMP2タンパク質が作られなくなり、ライソゾームの融合の機能に異常が生じ、オートファゴソームが異常に蓄積することにつながる可能性が示唆されています。ダノン病では、筋細胞内に異常に多数のオートファゴソーム(自己貪食空胞)蓄積が認められ、そのことがダノン病で見られる筋肉細胞の破壊につながる可能性があるとされています。
過剰自己貪食を伴うミオパチー(XMEA)では、X染色体のXq28領域に存在するVMA21遺伝子が原因遺伝子であることが明らかになっています。VMA21遺伝子は、液胞型ATPアーゼ集合因子21と呼ばれるタンパク質の設計図であり、このタンパク質はライソゾーム内の液性を酸性に保つために働くことが知られているため、ダノン病と同じようにライソゾームの正常な機能が失われると考えられますが、詳細なメカニズムはわかっていません。
その他、自己貪食空胞性ミオパチーに含まれる、乳児型自己貪食空胞性ミオパチー、成人発症型の(多臓器障害を伴う)自己貪食空胞性ミオパチーなどについては、原因となる遺伝子や、発症のメカニズムなどはわかっていません。
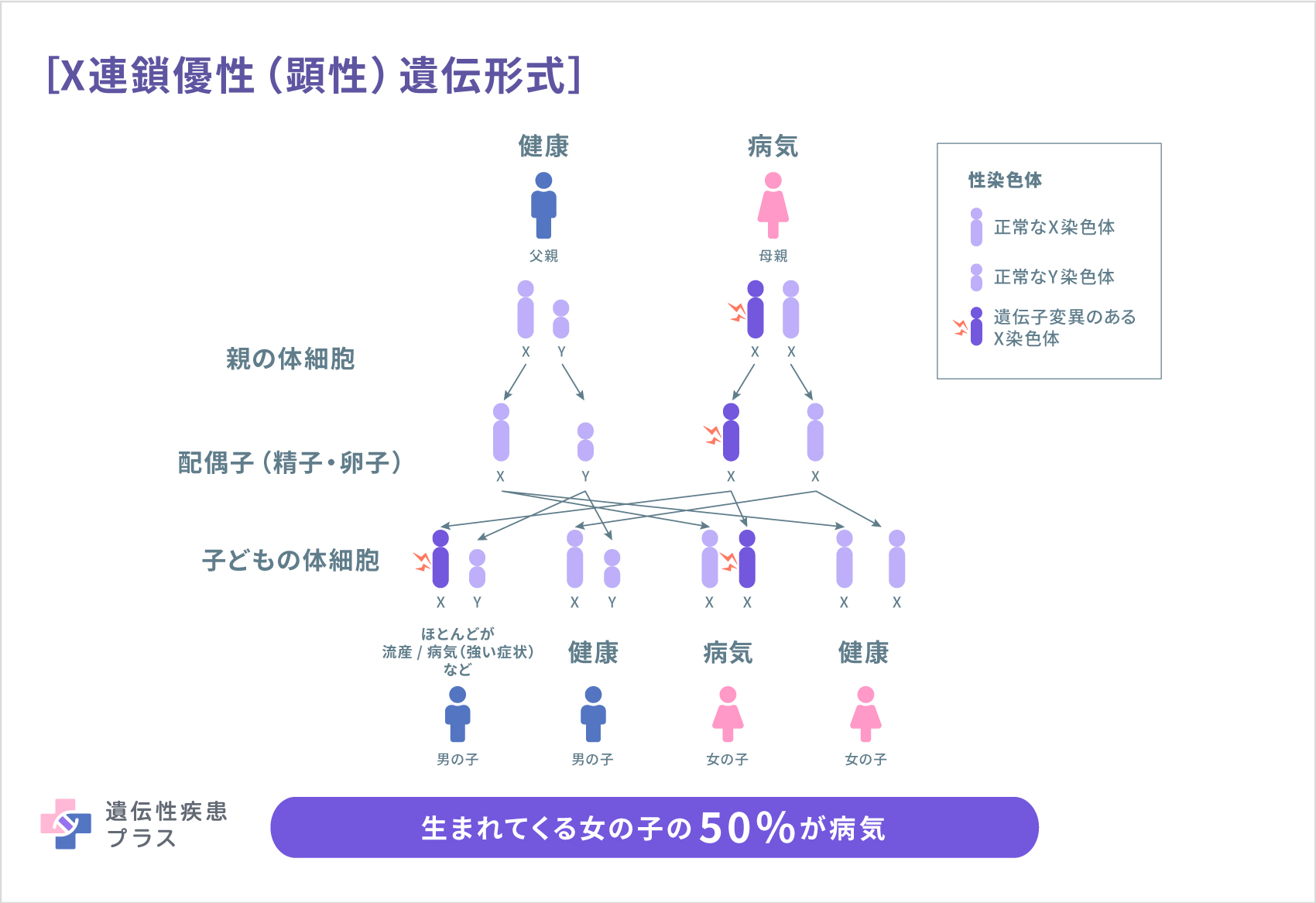
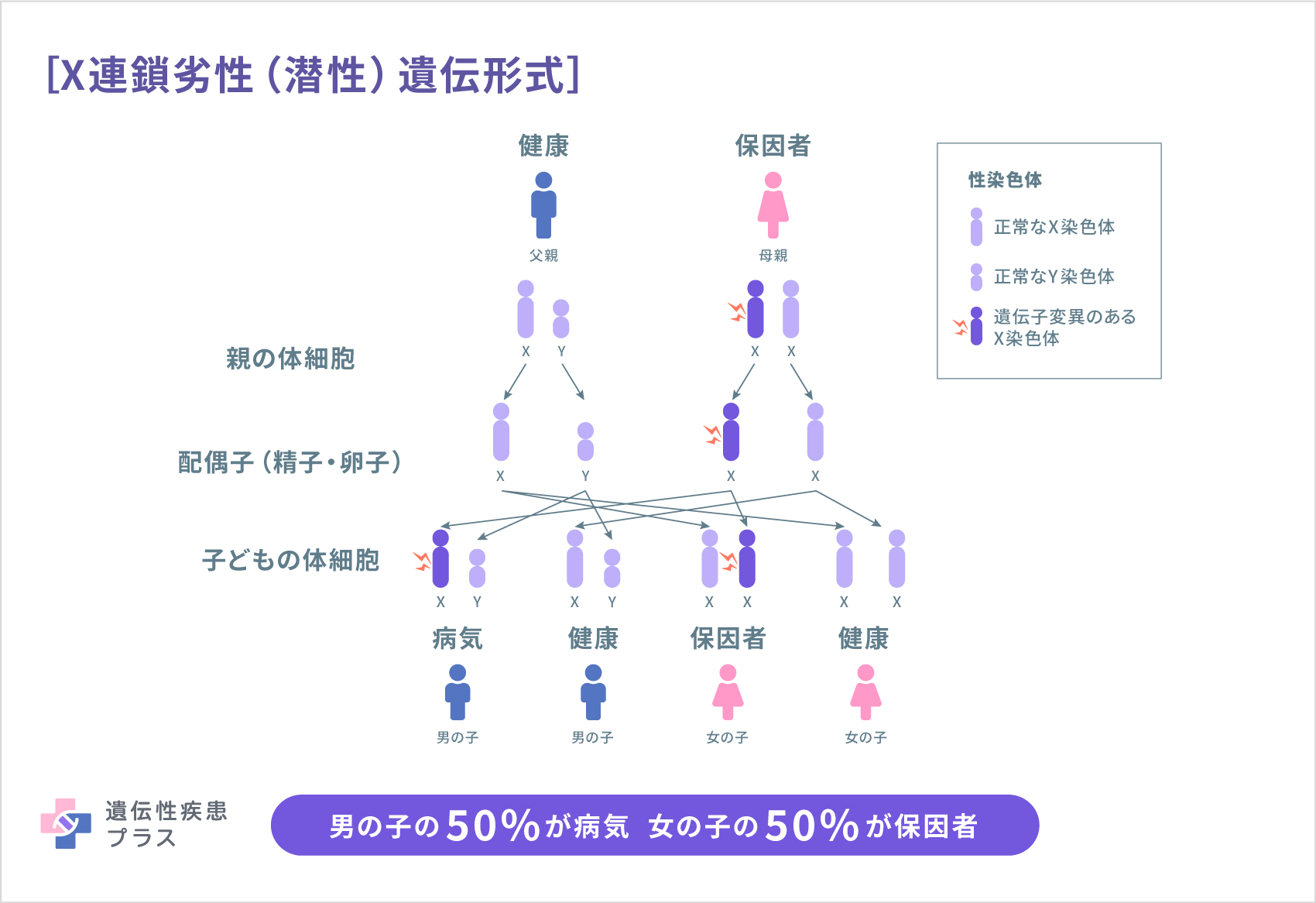
自己貪食空胞性ミオパチーに含まれる疾患のうち、ダノン病はX連鎖優性(顕性)遺伝形式、過剰自己貪食を伴うミオパチー(XMEA)はX連鎖劣性(潜性)遺伝形式と呼ばれる遺伝形式で遺伝します。
これらの遺伝形式は、どちらも性染色体であるX染色体が関連しています。ヒトが持つ性染色体にはX染色体とY染色体の2種類があり、XYの組み合わせは男性、XXの組み合わせは女性になります。
X連鎖優性(顕性)遺伝形式では、女性(XX)は変異のある遺伝子のコピーを1つ受け継ぐ事で発症します。男性(XY)の場合、X染色体を1つしか持たないため、ほとんどの場合、男性は女性よりもこの病気において重篤な症状が見られます。
X連鎖劣性(潜性)遺伝形式では、女性(XX)の場合は、変異のあるX染色体を持っていても、多くの場合は2本の染色体のうちもう一方の正常な遺伝子が機能を補うため、発症せず保因者となります。男性(XY)は、変異のある遺伝子のコピーを受け継ぐことで発症します。また、男性(父親)がこの変異を持つ場合には、息子には変異が受け継がれませんが、娘にはその変異のあるX染色体が受け継がれます。女性がこの変異を持つ場合には50%の確率でその原因遺伝子が存在するX染色体が娘または息子に受け継がれます。
どのように診断されるの?
自己貪食空胞性ミオパチーに含まれる疾患のうち、「ダノン病」「過剰自己貪食を伴うミオパチー(XMEA)」の診断は以下の基準で行われます。
これらの疾患の診断には、臨床的な症状だけではなく、筋肉の検査(筋病理所見)が重要になります。特に、自己貪食空胞性ミオパチーとは異なる性質の自己貪食空胞を有する糖原病2型(ポンペ病)、縁取り空胞を伴う遠位型ミオパチー、封入体筋炎などではないと診断されること(鑑別診断)も重要となります。
ダノン病の診断は、
A)臨床的な症状として、1)肥大型か拡張型心筋症、2)進行性の筋力低下および筋萎縮が見られること(男性のみ)
B)筋生検(筋肉の組織を採取して調べる検査)の結果、3)自己貪食空胞を伴う筋線維、4)空胞膜上でのアセチルコリンエステラーゼ活性(骨格筋での組織化学染色)が認められること
上記の、A)またはB)のどちらかを満たし、さらに、免疫組織化学染色かウェスタンブロット解析などでLAMP2タンパク質の欠損が認められるか、遺伝学的検査で、LAMP2遺伝子変異が認められることによって診断されます。LAMP2の異常が認められない場合においても、A)とB)の両方が認められた場合には、この病気の可能性があると診断されます。
過剰自己貪食を伴うミオパチー(XMEA)の診断は、
A)臨床的な症状として、緩徐進行性の筋力低下および筋萎縮
B)筋生検の結果、自己貪食空胞を伴う筋線維と空胞膜上でのアセチルコリンエステラーゼ活性(骨格筋での組織化学染色)が認められること
上記の、A)またはB)のどちらかを満たし、さらに、遺伝学的検査で、VMA21遺伝子変異が認められることによって診断されます。VMA21遺伝子変異が認められない場合においても、A)とB)の両方が認められた場合には、この病気の可能性があると診断されます。
どのような治療が行われるの?
どこで検査や治療が受けられるの?
日本で自己貪食空胞性ミオパチーやダノン病の診療を行っていることを公開している、主な施設は以下です。
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
難病の患者さん・ご家族、支えるさまざまな立場の方々とのネットワークづくりを行っている団体は、以下です。
参考サイト
- 難病情報センター
- 厚生労働科学研究費補助金 難治性疾患等政策研究事業「希少難治性筋疾患に関する調査研究」班編、自己貪食空胞性ミオパチー診療の手引き
- MedlinePlus
- Genetic and Rare Diseases Information Center
- Online Mendelian Inheritance in Man(R) (OMIM(R))
- GeneReviews
- orphanet
- NORD






