



どのような病気?
キャンポメリック ディスプラジアは、主に骨格、生殖器系、顔などに症状が現れる、先天性の希少遺伝性疾患です。重症度はさまざまですが、多くの場合重篤で、生後30日以内(新生児期)に命を落とします。
「キャンポメリック」はギリシャ語の「曲がった手足」を意味する言葉に由来しています。この疾患の特徴として、ほとんどの人で生まれたときから足の長骨(特に脛骨)が曲がっていることが挙げられます。腕が曲がっている人もいます。足の骨が曲がった部分には、特徴的な皮膚のくぼみが見られることがあります。その他の特徴として、短い足、股関節の脱臼、肩甲骨の発達不全、肋骨が11対(通常は12対)、首の骨の異常、内反足・外反足、頭蓋骨・骨盤の変形など、骨格の異常が見られます。この異常は骨軟骨の異形成によるもので、生まれる前に発症し始めるため、母親のおなかの超音波検査で見つかることもよくあります。肋骨が少なく、胸郭の発達が不十分なことによる呼吸窮迫や肺の成長への影響は、最も深刻な症状の1つです。
キャンポメリック ディスプラジアでは、典型的な男性の染色体パターン(46、XY)を有する人の約75%が、男女の区別がつきにくいあいまいな性器、または女性器を持っています。また、内部生殖器が外性器と一致しない場合があります。例えば、外性器が女性で、精巣を持っていたり、精巣と卵巣の両方を持っていたりすることがあります。
キャンポメリック ディスプラジアでは、小さなあご、眼球突出、平坦な顔など、特徴的な顔立ちも見られます。また、体の大きさに対し頭が大きいという特徴もあります。ピエール・ロバン・シーケンスと呼ばれる身体的特徴もよく見られます。ピエール・ロバン・シーケンスは、口蓋裂、舌根沈下(舌が喉の方に落ちている)、小顎症などを含みます。また、喉頭気管軟化症(上気道を形成する軟骨が弱い状態)で生まれることがよくあり、これが原因で気道が部分的に塞がれ、呼吸困難を起こすことがあります。こうしたことが、乳児の生存率低下の一因となっています。
この病気の人の10%程度は、新生児期に命を落とすことなく、生存します。乳児期を過ぎると、成長するにつれて脊柱側弯症、再発性呼吸器感染症、難聴、軽度~中等度の学習障害、低身長、股関節脱臼などの合併症を起こす可能性があります。
| キャンポメリック ディスプラジアで見られる症状 |
|---|
99~80%で見られる症状 肋骨が11組、口蓋裂、腓骨形成不全、平坦な顔、股関節脱臼、下前腸骨形成不全、喉頭軟化症、大頭症、小顎症、胸囲が小さい、頸椎の骨化不良、骨折しやすい、呼吸不全、脊柱側弯症、短い首、肩甲骨が小さい、脛の骨が曲がっている、気管支軟化症 |
79~30%で見られる症状 男女の区別があいまいな外性器、曲がった大腿骨、間隔の広い目、耳介低位、男性で内部生殖器が外性器と一致しない、眼球突出、低身長、皮膚のくぼみ、内反足 |
29~5%で見られる症状 心臓血管系の形態異常、嗅覚の異常、落ち込んだ鼻梁、聴覚障害、水腎症、脊柱後弯症、脳室拡大 |
割合は示されていないが見られる症状 心臓の異常、胸骨の骨化不全、無呼吸、短い眼瞼裂、手足が短く低身長、成長障害、筋緊張の低下、高い額、水頭症、頚椎の形成不全、腸骨稜の形成不全、肩甲骨の形成不全、後側弯症、羊水過多症、呼吸困難、性転換、手や足の指の骨が短い、小さな顔、細い肋骨、胸部形成不全、広い大泉門 |
米国希少疾病協議会(NORD)のウェブサイトによると、この疾患は、約100例が医学文献で報告されています。希少なため、疫学データは限られていますが、欧州の希少疾患情報サイト(orphanet)によると、生まれてくる子どもの約4万~8万人に1人がこの病気であると推定されています。日本の難病情報センターではおよそ3万人に1人、米国国立医学図書館のウェブサイト(MedlinePlus)ではおよそ4万~20万人に1人という推定値が掲載されています。また、NORDによると、この病気の女性は男性の約2倍いると考えられているものの、性別を誤って識別している場合もあるため、正しい数値ではない可能性もあります。
何の遺伝子が原因となるの?
キャンポメリック ディスプラジアの原因遺伝子として、17番染色体の17q24という位置に存在する「Sox9」遺伝子が見つかっています。Sox9遺伝子は、胚発生時に重要な役割を果たすSOX9タンパク質の設計図となるタンパク質です。SOX9タンパク質は、DNAの特定の領域にくっつくことで、そこにある遺伝子の働きを制御する、転写因子と呼ばれるものの仲間です。SOX9は多くの組織や器官の形成に関わっていますが、特に骨格や性決定に重要な遺伝子の活性を調節します。Sox9遺伝子内、またはその近傍の変異により、こうした遺伝子の働きが正しく制御されなくなることで、この病気が発症すると考えられています。
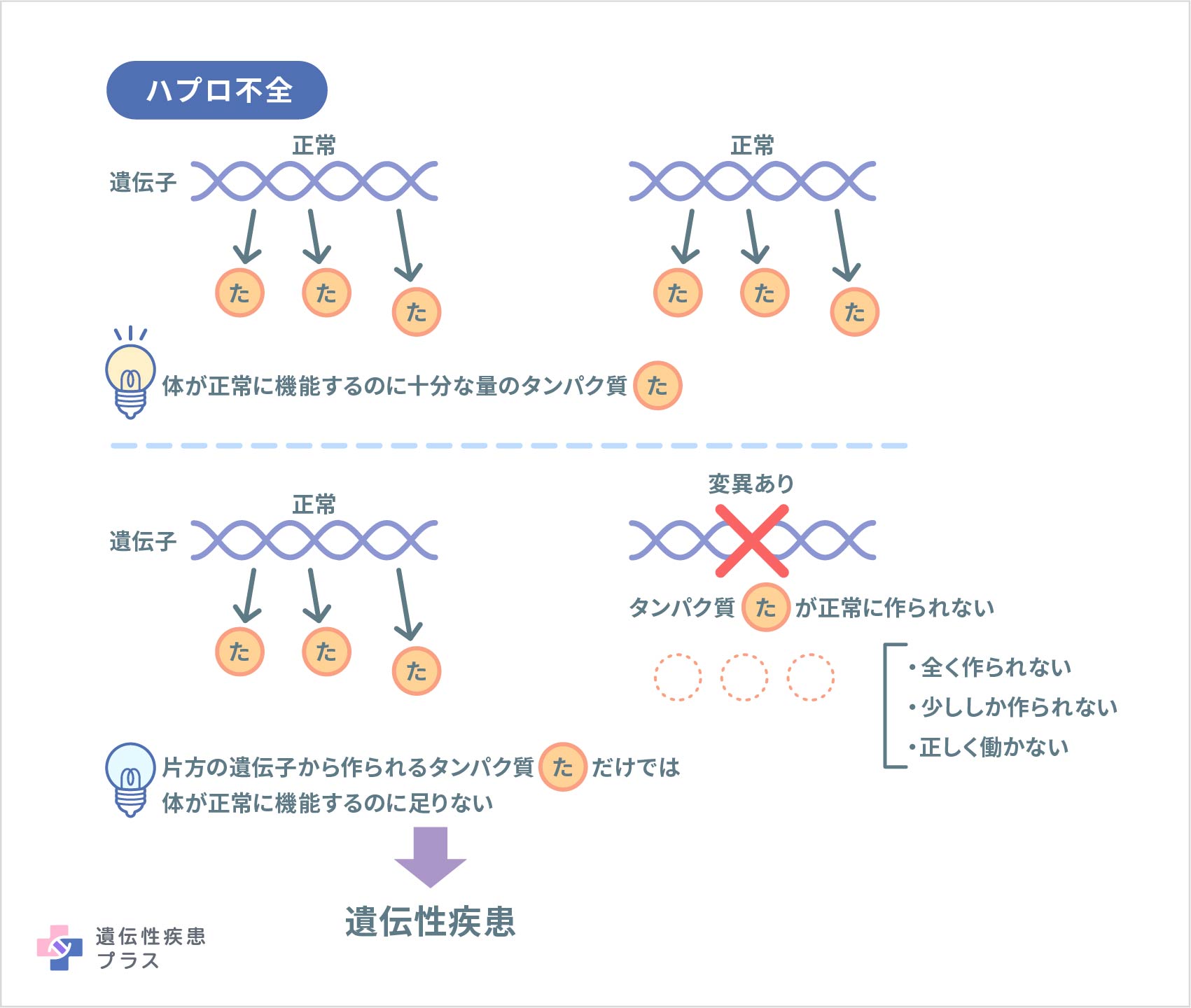
キャンポメリック ディスプラジアのほとんどの症例は、Sox9遺伝子内の変異により、SOX9タンパク質が必要な量作られない、もしくは、正しく機能するSOX9タンパク質が作られない状態にあります(ハプロ不全)。症例の約5%は、Sox9遺伝子の近傍に染色体異常(転座)が見られます。この場合、Sox9遺伝子内に変異がある症例よりも軽症となる傾向があります。染色体異常が起きている部分は、Sox9遺伝子の働きを調節するDNA領域であるため、この異常によりSOX9遺伝子の正しい働きが妨げられ、発症すると考えられています。
キャンポメリック ディスプラジアの遺伝的変異は、大部分が両親では変異がなく、子どもで新たに発生するものです(新生変異による孤発例)。したがって、この病気の「家系」がみられる例はほとんどありません。両親のどちらかがキャンポメリック ディスプラジアであった場合は、常染色体優性(顕性)遺伝形式で遺伝するため、親から子へは50%の確率で遺伝します。また、両親ともに症状がなく、生まれた子どもがきょうだいでキャンポメリック ディスプラジアを発症したという家族の報告がいくつかあります。この原因として、両親のどちらかで、正常な生殖細胞(精子または卵子)とSox9遺伝子に変異を持つ生殖細胞が混在している(性腺モザイク)可能性が考えられています。

どのように診断されるの?
日本では現在、「キャンポメリック ディスプラジアの臨床情報調査と病因・病態の研究班」が、診断基準の策定を含め、研究を進めています。
米国希少疾病協議会(NORD)の情報によると、診断は、身体的所見、X線検査(脊椎、股関節、胸部、脚など)、腎臓の超音波検査、心臓の心エコー検査などに基づき行われます。採血をして、その血液を用いて遺伝子検査を行うことで、Sox9遺伝子の変異を確認する場合もあります。
欧州の希少疾患情報サイト(orphanet)によると、キャンポメリック ディスプラジアと似た症状があり、鑑別診断が必要な疾患として、出生前では骨形成不全症2型および3型、低ホスファターゼ症、タナトフォリック骨異形成症、シュワルツ・ヤンペル症候群、出生後では先天性脊椎骨端異形成症(SEDC)、捻曲性骨異形成症、ラーセン症候群が挙げられています。
どのような治療が行われるの?
今のところ、キャンポメリック ディスプラジアを根本的に、つまり、遺伝子から治すような治療法は見つかっていません。骨の変形などに対し有効な、根本的な治療法もありません。比較的軽症の人は、生涯にわたり、診療を受けていく場合もあります。
治療は、それぞれの症状に対して行われます。深刻な骨の変形に対しては、手術が行われることもあります。聴覚障害には補聴器が使用されます。胸郭を保って肺の成長や呼吸を助けるために、チタン製の体内埋め込み型の医療機器が用いられる場合もあります。
男性型の染色体を持ち、性器の男性化が不十分な人では、性腺芽細胞腫のリスクが高いため、性腺の切除が検討される場合があります。その他、性別に関わる症状などに対し、遺伝カウンセリングを受けることが患者さんやご家族にとって有益となる場合があります。
どこで検査や治療が受けられるの?
日本でキャンポメリック ディスプラジアの診療を行っていることを公開している、主な施設は以下です。
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
難病の患者さん・ご家族、支えるさまざまな立場の方々とのネットワークづくりを行っている団体は、以下です。





