



どのような病気?
CDKL5欠損症は、乳児期の早くに始まるてんかん発作、発育の遅れ、知的障害などを特徴とする遺伝性疾患です。CDKL5欠損症のてんかん発作は、生後3か月以内に始まることが多くありますが、生後1週間以内に始まることもあります。このてんかんは薬でコントロールすることが難しく、多くの人で、1日に1~5回の発作が起こります。てんかん発作のパターンは年齢とともに変化し、3つのステージに分けられています。発症時~10週後くらいまで(ステージ1)は、焦点性発作(大脳の一部に起こる発作)や全身性強直間代発作(典型的なてんかん発作)を頻回にくり返しますが、多くの場合、発作が起きていないときの脳波(間欠期脳波)には異常がみられません。その後、3歳くらいまでに点頭てんかん(ウエスト症候群)と呼ばれるタイプの、ヒプスアリスミアという脳波がみられる発作が起こるようになり(ステージ2)、さらに年齢が進むと強直発作やミオクロニー発作と呼ばれる発作が主に起こり、間欠期脳波に「多焦点性棘徐波複合」と呼ばれる状態がみられる難治性てんかんになっていきます(ステージ3)。てんかん発作は、成人になるまでに約半数で起こらなくなりますが、残りの半数は、薬でコントロールできない発作が続きます。
この病気では、乳児期の早くに明らかな筋緊張低下と精神運動発達遅滞が認められます。こうした症状は、座る、立つ、歩くなどの行動にも影響しますが、約3分の1は、支えなしに歩くことができるようになります。指で小さな物をつまむなどの細かい作業は難しい場合が多いものの、約半数は、自分の意思で手を使用することができるようになります。
CDKL5欠損症では言葉の発達も遅れ、ほとんどの人は、言葉を話せません。また、この病気のほとんどの人は、視力の障害を持っています。その他、睡眠障害、摂食障害、常同運動(拍手、手なめを繰り返すなど)、歯ぎしり、消化器系の症状(便秘、胃食道逆流症、空気の飲み込みなど)も見られる場合があります。
| CDKL5欠損症で見られる症状 |
|---|
| てんかん発作、特徴的な脳波、上向きの鼻、広い額、突き出た額、脳性視覚障害、便秘、目が大きく奥まっている、発達遅滞、てんかん脳症、胃食道逆流症、筋緊張低下、ミオクローヌス発作、発育遅延、過呼吸、歩けない、点頭てんかん、知的障害(IQ20未満)、多発性発作、アイコンタクトに乏しい、進行性の小頭症、脊柱側弯症、短い足、小さな手、常同運動、細い指先、下唇が厚い、上あごが高い、間隔の広い歯 |
症状は人によってさまざまで、てんかん発作がコントロールできて歩くこともできるようになる人から、難治性てんかんを伴った重度の精神運動発達遅滞となる人まで、いろいろです。
CDKL5欠損症は、自閉症状、手の常同運動、睡眠障害、呼吸障害(過換気・呼吸停止が起こる等)など、レット症候群と似た症状が現れることもあるため、以前は非典型的レット症候群(レット症候群の非典型例)として説明されていました。しかし、CDKL5欠損症は、明らかな退行(これまでできていたことができなくなる状況)が見られないなど、レット症候群と異なる部分があることなどから、現在は別の疾患と考えられるようになっています。今のところ日本では、CDKL5欠損症は、レット症候群の非典型例として、国の指定難病対象疾患(指定難病156)、および、小児慢性特定疾病の対象疾患となっています。
厚生労働省の資料によると、日本にいるCDKL5欠損症の患者さんは、100人未満とされています。また、米国国立衛生研究所の疾患情報サイト「MedlinePlus」によると、CDKL5欠損症は、4万〜6万人に1人の頻度で生まれるとされており、国際CDKL5研究財団(International Foundation for CDKL5 Research;IFCR)のサイトでは、「(世界で)毎週少なくとも2人がCDKL5と診断されている」と表現されています。患者さんは男女ともにいますが、その遺伝形式から、診断された人の約90%は女性であることがわかっています。
また、CDKL5欠損症の人たちの平均余命や診断される年齢について、IFCRのウェブサイトでは、「CDKL5欠損症は2004年に発見されたばかりの病気なのでまだ十分なデータが得られていない」としたうえで、40歳以上の患者さんについての論文が報告されていること、10~30歳代の患者さんも多くいること、乳幼児で診断がつくケースがほとんどだが40~50歳代で診断される人も出てきていることが、記されています。
何の遺伝子が原因となるの?
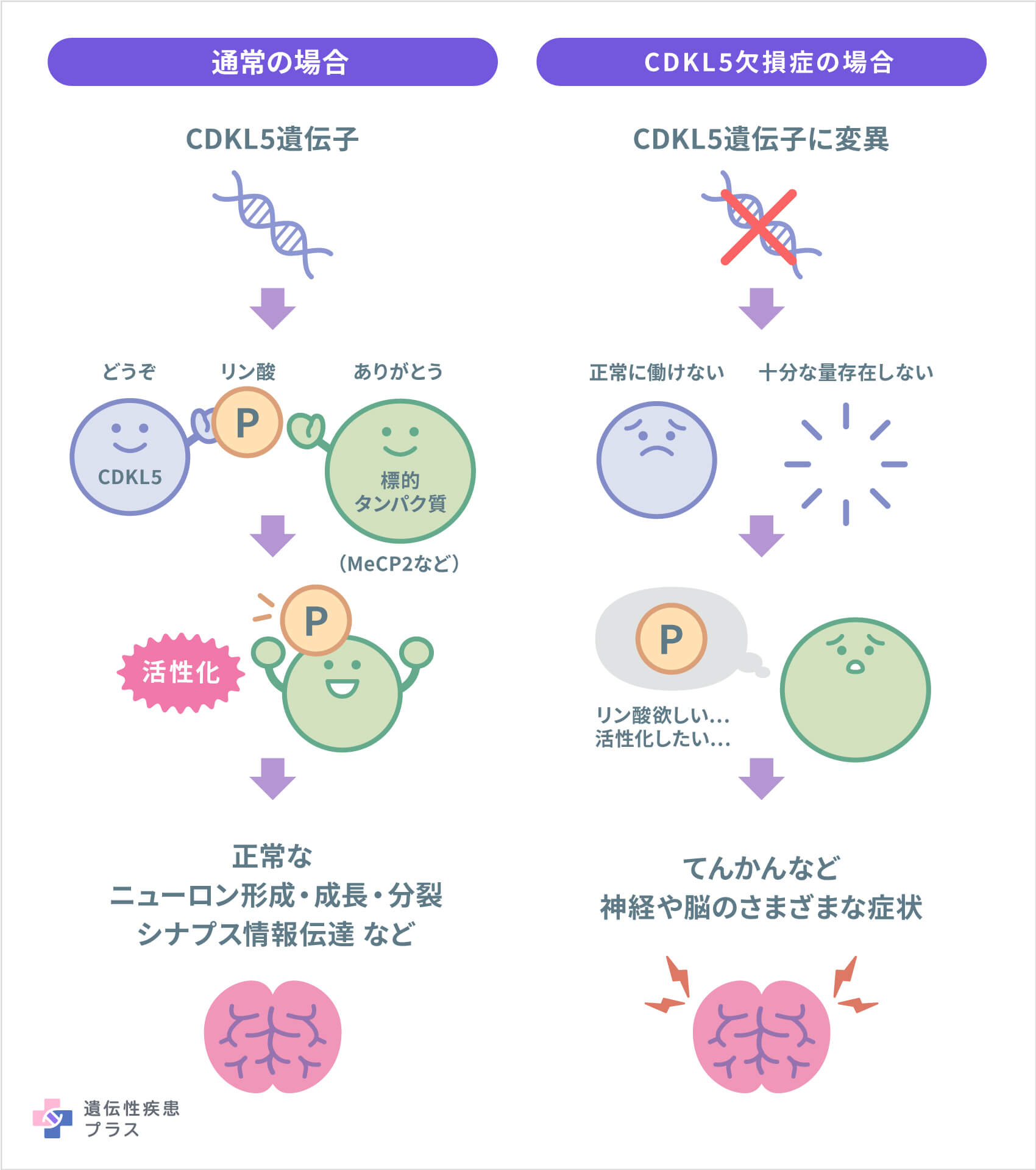
CDKL5欠損症は、X染色体のXp22.13という位置に存在するCDKL5遺伝子の異常によって引き起こされます。この遺伝子は、全身の細胞に存在するCDKL5(サイクリン依存性キナーゼ様5)というタンパク質(酵素)の設計図となる遺伝子です。CDKL5は脳で最も活発に働いており、この働きが正常な脳の発達と機能に不可欠であることがわかっています。具体的には、CDKL5が神経細胞(ニューロン)の形成、成長、分裂、シナプスによる情報伝達などに関与していることが、これまでの研究により示されています。
CDKL5は、他のタンパク質に「リン酸化」という化学変化を起こし、そのタンパク質の活性を変化させる、「キナーゼ」という種類の酵素です。CDKL5がリン酸化するタンパク質は、まだ全てがわかっているわけではありませんが、MeCP2タンパク質はそのうちの1つであるとわかっています。MeCP2は、ニューロンや脳細胞の機能や維持に重要な役割を果たすタンパク質の1つで、MeCP2遺伝子の異常は、レット症候群の原因です。
CDKL5遺伝子に変異があると、正常に働くCDKL5の量が減り、その結果、神経や脳に関するさまざまな症状が起こると考えられています。しかし、CDKL5欠損症の各症状をどのように引き起こすのか、そのメカニズムはまだ解明されていません。
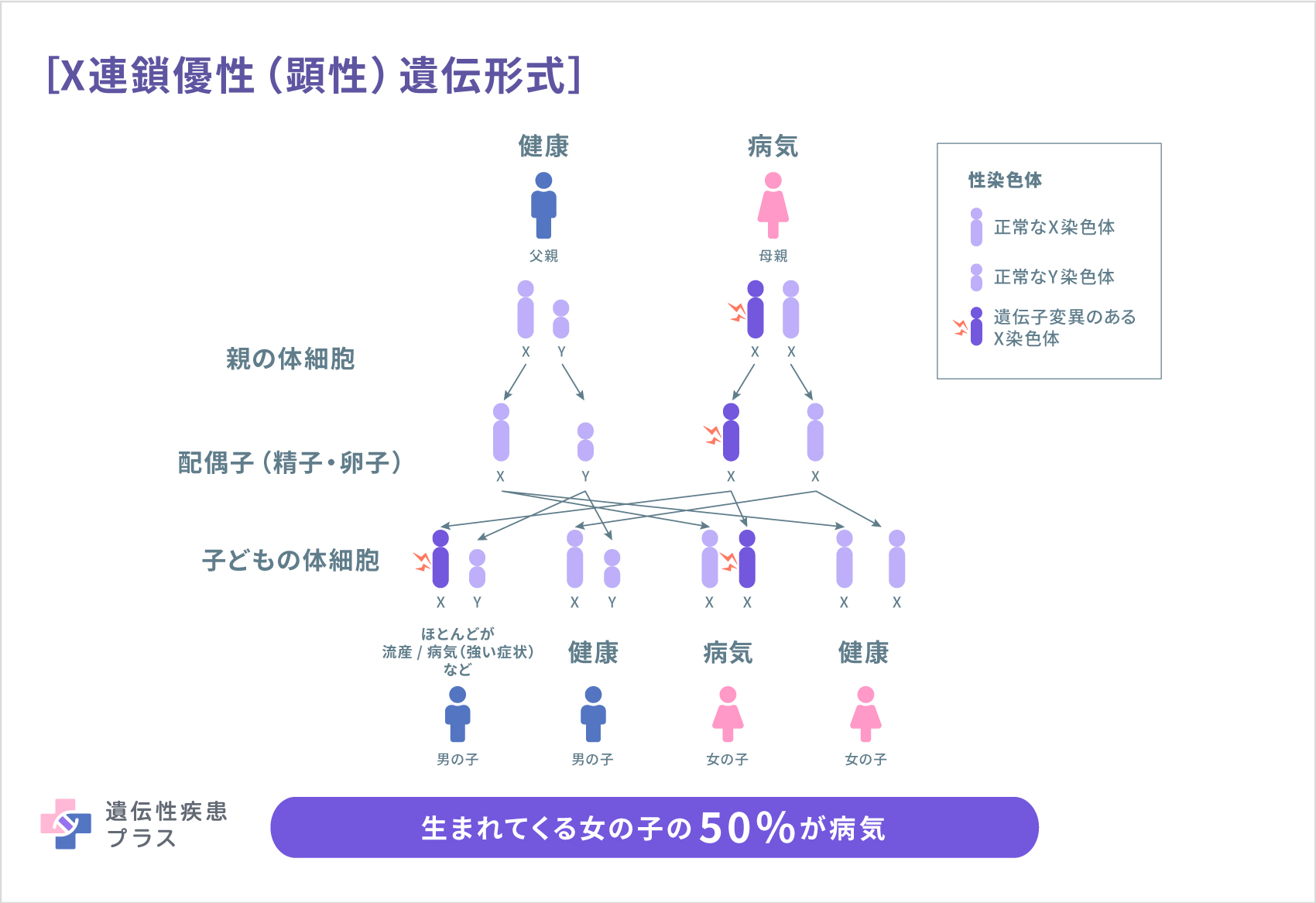
CDKL5欠損症は、X連鎖優性(顕性)遺伝形式で、親から子へ遺伝します。人間は、性染色体と呼ばれる染色体を2本持っています。性染色体にはX染色体とY染色体の2種類があり、XYの組み合わせは男性、XXの組み合わせは女性になります。CDKL5遺伝子は、X染色体に存在する遺伝子です。
男性が1本のみ有するX染色体のCDKL5遺伝子に変異があると、正常なCDKL5タンパク質が全く作られなくなる、もしくは生きていくのに必要な量作られなくなるため、多くの場合、生まれることなく流産となります。
しかし女性の場合は、X染色体を2本持っているので、1本が異常でも、もう1本のCDKL5遺伝子が正常に働き、機能を補完します。そのため、流産とならずCDKL5欠損症として生まれてきます。したがって、CDKL5欠損症は、一般に母親から娘に遺伝する形か、新生変異(その子どもで新たに起きた変異)による孤発例となりますが、CDKL5欠損症の遺伝子変異は、大部分が両親には変異がなく新生変異で、「家系」がみられる例はほとんどありません。ただし、1家系で3人のきょうだいがCDKL5欠損症を発症したという報告があり、この報告から、精子や卵子の前駆細胞である生殖細胞が「モザイク」という状態(CDKL52遺伝子変異をもつものともたないものが混在している)になっている親から遺伝する可能性があると指摘されています。
CDKL5欠損症の患者さんが子どもを持った場合には、母親から娘にこの病気が遺伝する確率は、50%です。つまり、CDKL5欠損症の母親から生まれる子どもは、CDKL5欠損症の女児:CDKL5欠損症でない女児:CDKL5欠損症でない男児=1:1:1となります。
また、CDKL5欠損症の重症度の違いには、「X染色体不活性化」と呼ばれる現象が関わっていると、研究者は考えています。女性はX染色体を2本持っているので、X染色体上の遺伝子が男性に比べ過剰に働き過ぎないように、生まれてくる初期の過程で2本のうちの1本が体細胞(卵子と精子細胞以外の細胞)でランダムに、永久に不活性化されます。これをX染色体不活性化といいます。しかし、この不活性化は変異のあるX染色体と変異のないX染色体に対して正確に50%ずつ全身で起こるわけではないため、脳内のニューロンで起こるX染色体不活性化の割合の偏りが、重症度に関わると考えられているのです。
どのように診断されるの?
厚生労働省の「CDKL5遺伝子関連てんかん」に関する資料に、「稀少てんかんに関する調査研究班」が作成した、CDKL5遺伝子関連てんかんの診断基準が記載されています。その内容は、下記Aの症状およびBの3からCDKL5遺伝子関連てんかんの疑いとなり、Dの遺伝学的検査で異常が認められた場合、診断確定となる、というものです。
A 症状
- 新生児期〜乳児期早期に難治性てんかんを発症
- 初発時の発作は焦点性発作、または全身強直間代発作
- 乳児期早期からの筋緊張低下と精神運動発達遅滞
- のちにWest症候群へと変容する
- 女児(まれに男児)
- 自閉症状
- 手の常同運動
B 検査所見
- 血液・生化学的検査所見:特異的所見なし
- 画像検査所見:特異的所見なし
- 生理学的所見:脳波(間欠期脳波で発症初期に異常がないかあっても軽度、その後ヒプスアリスミアを経て多焦点性へと変容していく)
C 鑑別診断
CDKL5遺伝子に病的な異常が同定されれば診断は確実となる。ただし臨床的にはMECP2遺伝子変異による古典的レット症候群やその他の遺伝子異常・代謝異常・器質的異常に起因するウエスト症候群などが鑑別となる
D 遺伝学的検査
CDKL5遺伝子の変異
どのような治療が行われるの?
今のところ、CDKL5欠損症を根本的に、つまり、遺伝子から治すような治療法は見つかっていません。そのため、現時点ではその人の症状に合わせて対症療法が行われます。
てんかん発作に対しては発作型に対応した抗てんかん薬が用いられます。しかし、現段階では、薬が有効でない場合が多くあります。ウエスト症候群をはじめとしたてんかんに対して、注射による治療である「ACTH療法」が行われる場合もあります。また、てんかんに対する食事療法である「ケトン食療法」が行われる場合もあります。
その他、運動や姿勢の異常に対してリハビリテーションが、知的障害・発達障害に対して療育などが考慮されます。
どこで検査や治療が受けられるの?
日本でCDKL5欠損症の診療を行っていることを公開している、主な施設は以下です。
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
CDKL5欠損症の患者会で、ホームページを公開しているところは、以下です。
参考サイト
- 難病情報センター
- 小児慢性特定疾病情報センター レット(Rett)症候群
- 厚生労働省 CDKL5遺伝子関連てんかん 資料1
- MedlinePlus
- Genetic and Rare Diseases Information Center
- Online Mendelian Inheritance in Man(R) (OMIM(R))
- International Foundation for CDKL5 Research






