



どのような病気?
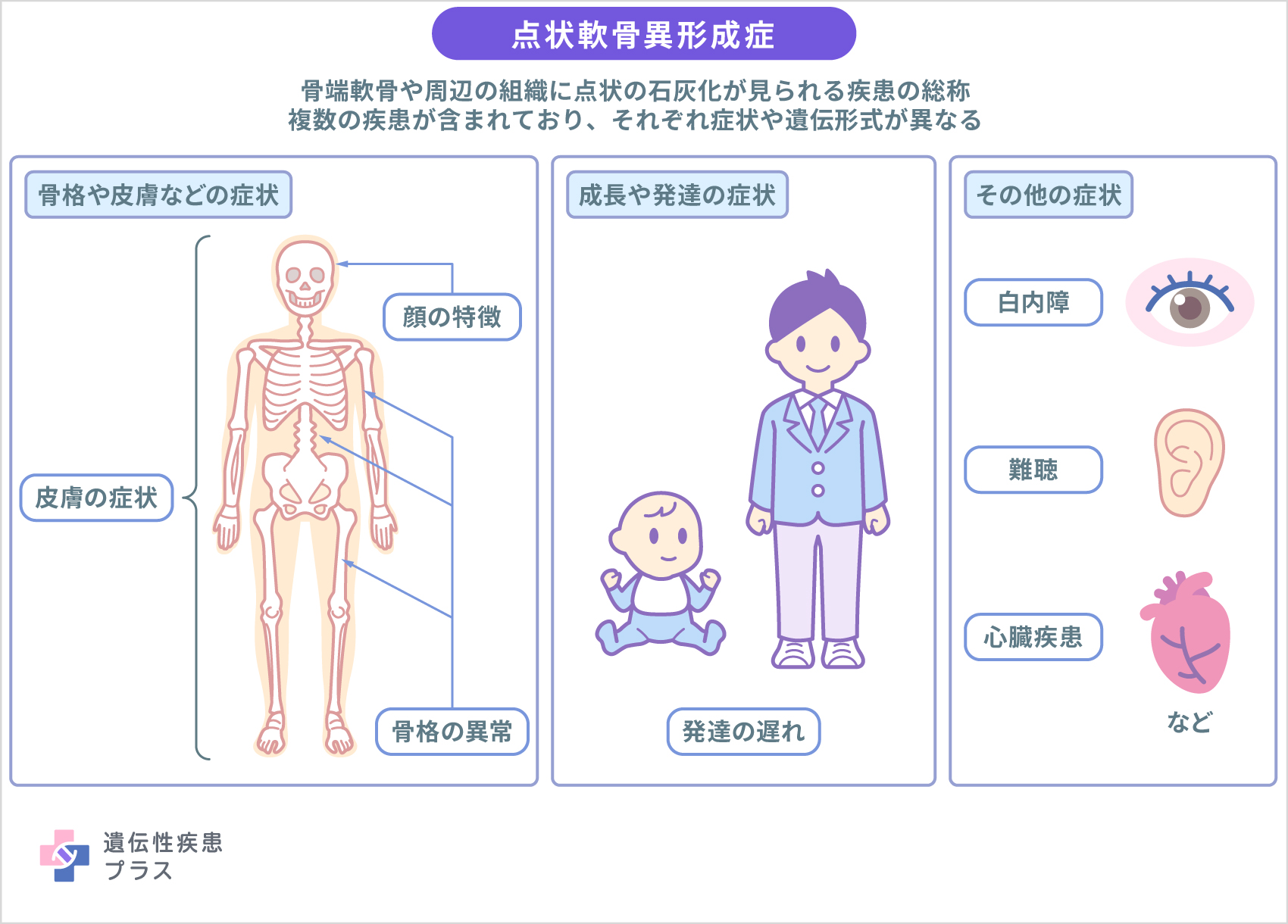
点状軟骨異形成症は骨端軟骨とその周囲の軟部組織に点状の石灰化を生じる疾患群の総称で、多くの疾患が含まれます。点状軟骨異形成症には、X連鎖性劣性末節骨短縮型点状軟骨異形成症(CDPX1)、X染色体優性Conradi-Hünermann型点状軟骨異形成症(CDPX2)、CHILD症候群、Keutel症候群、Greenberg骨異形成症、近位肢型点状軟骨異形成症、脛骨・中手骨型点状軟骨異形成症、Astley-Kendall骨異形成症が含まれます。また、点状軟骨石灰化は、ワルファリン胎芽症、ビタミンK欠乏症、全身性エリテマトーデス(SLE)胎芽症などの遺伝性疾患ではない病気においても見られることがあります。この記事では、ペルオキシソーム病に含まれる近位肢型点状軟骨異形成症、遺伝性ではない疾患以外の8つの病型について、点状軟骨異形成症として取り扱います。
X連鎖性劣性末節骨短縮型点状軟骨異形成症(CDPX1)は、重度の鼻骨低形成、低身長、遠位指節骨(指の第一関節より上の骨)低形成、特徴的な顔立ちなどが見られ、ほとんどの場合男性で発症します。多くの場合、学習能力や寿命に影響はありませんが、気道を形成する軟骨が異常に厚くなることにより呼吸が制限されるなどの重篤な合併症が引き起こされる場合もあります。また、首の脊椎骨の異常により脊髄が圧迫され、痛み、しびれ、脱力などが生じることもあります。まれに、発達の遅れ、難聴、視覚異常、心臓疾患などが見られる場合もあります。
<CDPX1で見られる症状>
点状軟骨異形成、鼻骨上顎低形成、趾の末節骨短小(足の指先の骨の短小)、低身長、呼吸器異常、聴覚障害、頸椎異常、発達遅滞、認知機能発達遅延
X染色体優性Conradi-Hünermann型点状軟骨異形成症(CDPX2)は、ほとんどの場合女性で発症します。この病型において、症状や重症度は患者さんごとに大きく異なり、ほとんど症状が見られない人から胎児死亡や重度の成長遅延が起こる場合もあるなど多岐にわたります。家族内や同じ遺伝子変異を持つ場合でも重症度が異なることがあります。CDPX2で見られる点状軟骨異形成症は、腕や脚の長骨、肋骨、脊椎などの骨格や、気管を構成する軟骨などに影響を及ぼし、上腕骨や大腿骨の骨の短縮や骨の長さが左右で異なる、脊椎の異常な湾曲(脊柱後側弯症)も見られ、その結果として低身長となる傾向があります。その他に、顔と頭部の非対称、乳児期における魚鱗癬(乾燥した鱗状の皮膚の斑点)、まばらで粗い頭髪、白内障などの眼の異常なども見られることがあります。また、まれに男性でCDPX2を発症することがあり、その場合女性よりも重篤な症状となることが報告されています。男性の場合上記の症状に加えて、筋緊張低下、脳構造の変化、中程度から重度の発達遅延、てんかん発作、特徴的な顔立ち、その他の先天性欠損などが見られ、命に関わる症状となる場合も多いとされます。CDPX2は、発見した医師の名前から、Conradi-Hünermann症候群やHapple症候群、Conradi-Hünermann-Happle症候群と呼ばれることもあります。多くの場合、CDPX2の患者さんの寿命は平均的ですが、重度の脊柱側弯症により心臓や肺の機能が損なわれることで悪影響を与える可能性もあるとされます。
<CDPX2で見られる症状>
発育不全、低身長、前頭隆起、低い鼻梁(低いはなすじ)、まばらな眉毛、骨格異常、点状軟骨異形成、側弯症(背骨の湾曲)、多趾症(足の指が多いこと)、皮膚・髪・爪の異常、眼疾患(白内障、小眼球症)
CHILD症候群は、魚鱗癬状紅斑(うろこ状の紅斑)、先天性片側異形成、四肢欠損を特徴とした病型です。この病型の症状や程度は人によって異なりますが、通常は体の片側だけに症状が現れることが多く、体の左側よりも右側に症状が出やすいとされています。この病気で見られる魚鱗癬状紅斑は、皮膚のひだやシワに発生する可能性が高く、通常は顔に現れません。皮膚の異常は、出生時に存在するか生後数週間以内に現れますが、時間の経過とともに改善する可能性があります。発達の早い時期に腕や脚の形成が妨げられ、手足の指の骨が短くなる、あるいは、手足が短くなったり欠損したりする場合もあります。手足の異常と皮膚の異常は、体の同じ側に現れることが多いとされます。脊椎側弯症や関節の変形(拘縮)のほか、脳、心臓、肺、腎臓の発達に影響が見られる場合もあります。男性の場合には出生前に死亡するリスクが高いとされています。
Keutel症候群は石灰化異常による骨格形態異常、心血管系の異常、聴覚障害、発達遅滞を特徴とした病型です。この病型で見られる石灰化異常は、耳、鼻、喉頭、気管支樹の軟骨のほか、長骨の骨端線の点状化、脊柱椎骨などにも見られます。上顎や顔面中央の形成不全(鼻すじが広く陥没し、鼻翼が小さい)などの顔立ちの特徴も見られることがあります。また、難聴や繰り返す中耳炎および/または副鼻腔炎、てんかん発作、軽度の知的障害、呼吸器感染症が頻繁に起こる、鼻声、低身長なども認められます。血管石灰化や脳石灰化、モヤモヤ病、動脈狭窄なども起こる場合があるとされています。
Greenberg骨異形成症は、胎児水腫、四肢短縮、軟骨石灰化異常を特徴とする非常にまれな軟骨形成異常症です。この病型では、骨が適切に発達せず、虫食い骨と呼ばれる特徴的な斑点状の外観がX線画像で確認されます。胎児期に子宮内で亡くなってしまうことも多い病型です。
脛骨・中手骨型点状軟骨異形成症は、不均衡に短い中手骨(手のひらの骨)と脛骨(けいこつ・すねの骨)を特徴とし、重度の低身長、上肢と下肢の左右両側の短縮、特徴的な顔立ち(平坦な中顔面と鼻)などが認められます。まれに、新生児期の頻呼吸、水頭症、軽度の発達遅延が合併することもあります。その他の骨の異常として、湾曲した長骨、扁平脊椎、椎間裂なども含まれることがあります。白内障や皮膚の異常は通常見られません。この病型の予後は良好とされています。
Astley-Kendall骨異形成症は頭蓋骨や体軸に関わる骨や手根骨の形成不全を生じる病型です。Greenberg骨異形成症と同様、胎児期に子宮内で亡くなってしまう場合も多いとされています。
点状軟骨異形成症全体の発症頻度は不明です。海外の報告では、点状軟骨異形成症のうちCDPX1は50万人に1人、CDPX2は10~20万人に1人とされています。日本の点状軟骨異形成症の患者数は不明です。日本整形外科学会骨系統疾患全国患者登録では1990~2015年の26年間に64例が登録されています。
点状軟骨異形成症は、「点状軟骨異形成症(ペルオキシソーム病を除く。)」として、小児慢性特定疾病の対象となっています。また、CDPX2(Conradi-Hünermann-Happle症候群)とCHILD症候群は「先天性魚鱗癬」の一病型として指定難病対象疾病に含まれる場合があります。
何の遺伝子が原因となるの?
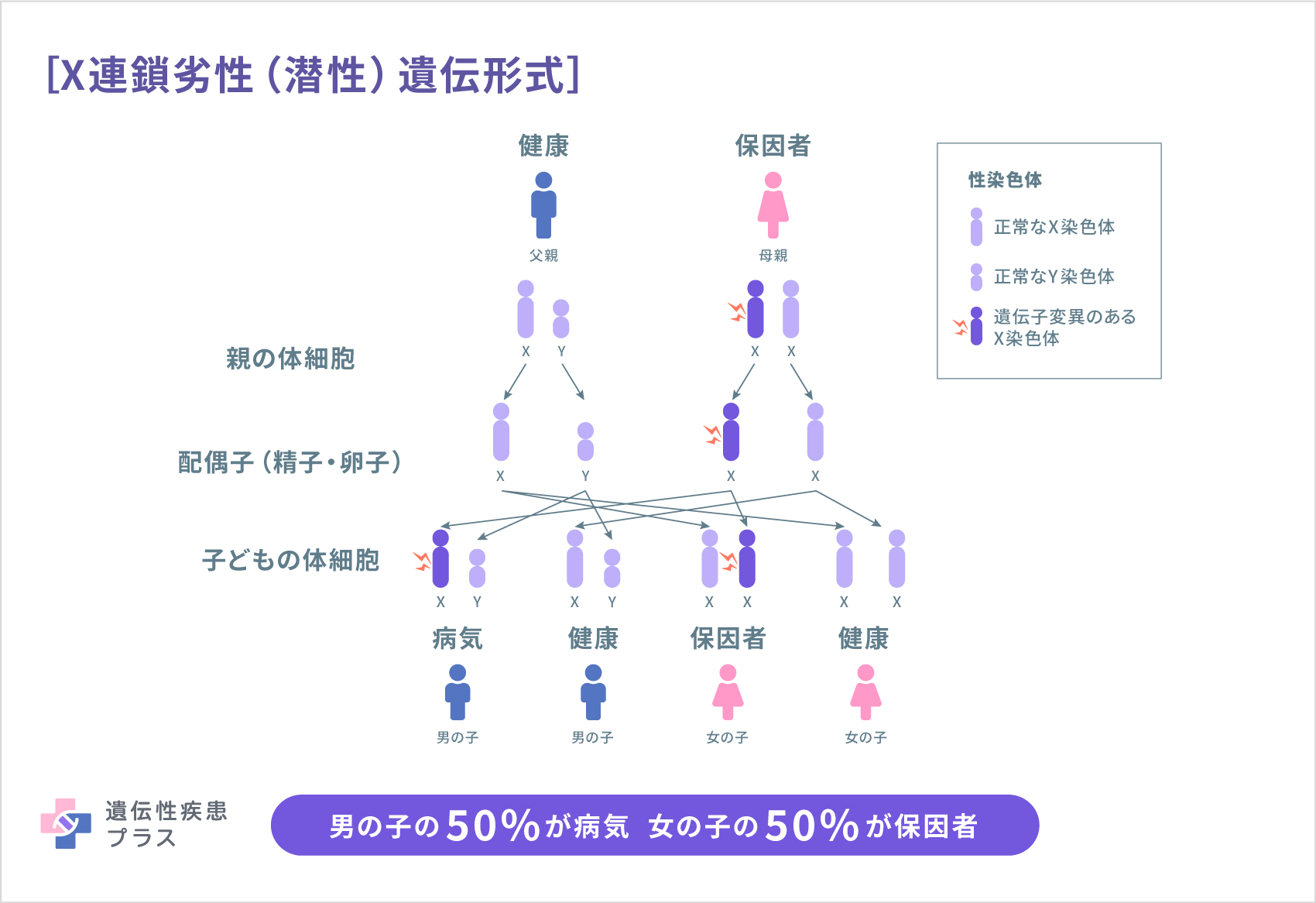
CDPX1ではX染色体上にあるARSL遺伝子の変異、もしくはARSL遺伝子を含むX染色体の一部の領域の欠失が報告されています。ARSL遺伝子はアリルスルファターゼLという酵素の産生に関与しています。この酵素の機能についての詳細はわかっておらず、ARSL遺伝子の変異によるスルファターゼLの欠損がどのようにCDPX1の発症に関与するのかはまだはっきりわかっていませんが、アリルスルファターゼLは、骨の成長や骨密度の維持に重要と考えられている、ビタミンKに関わる経路に関与していると考えられています。
CDPX1はX連鎖劣性(潜性)遺伝形式で遺伝します。ARSL遺伝子はX染色体にあり、母親がARSL遺伝子の変異を有する場合、子どもは50%の確率で変異したARSL遺伝子を受け継ぎます。子どもが男児の場合、X染色体は1つしかないため、男児は変異したARSL遺伝子の影響を受けます。子どもが女児で変異した遺伝子を受け継いだ場合には保因者となります。CDPX1の男性の患者さんではその子どもが女児の場合、変異したARSL遺伝子が受け継がれますが、子どもが男児の場合にはARSL遺伝子変異は受け継がれません。
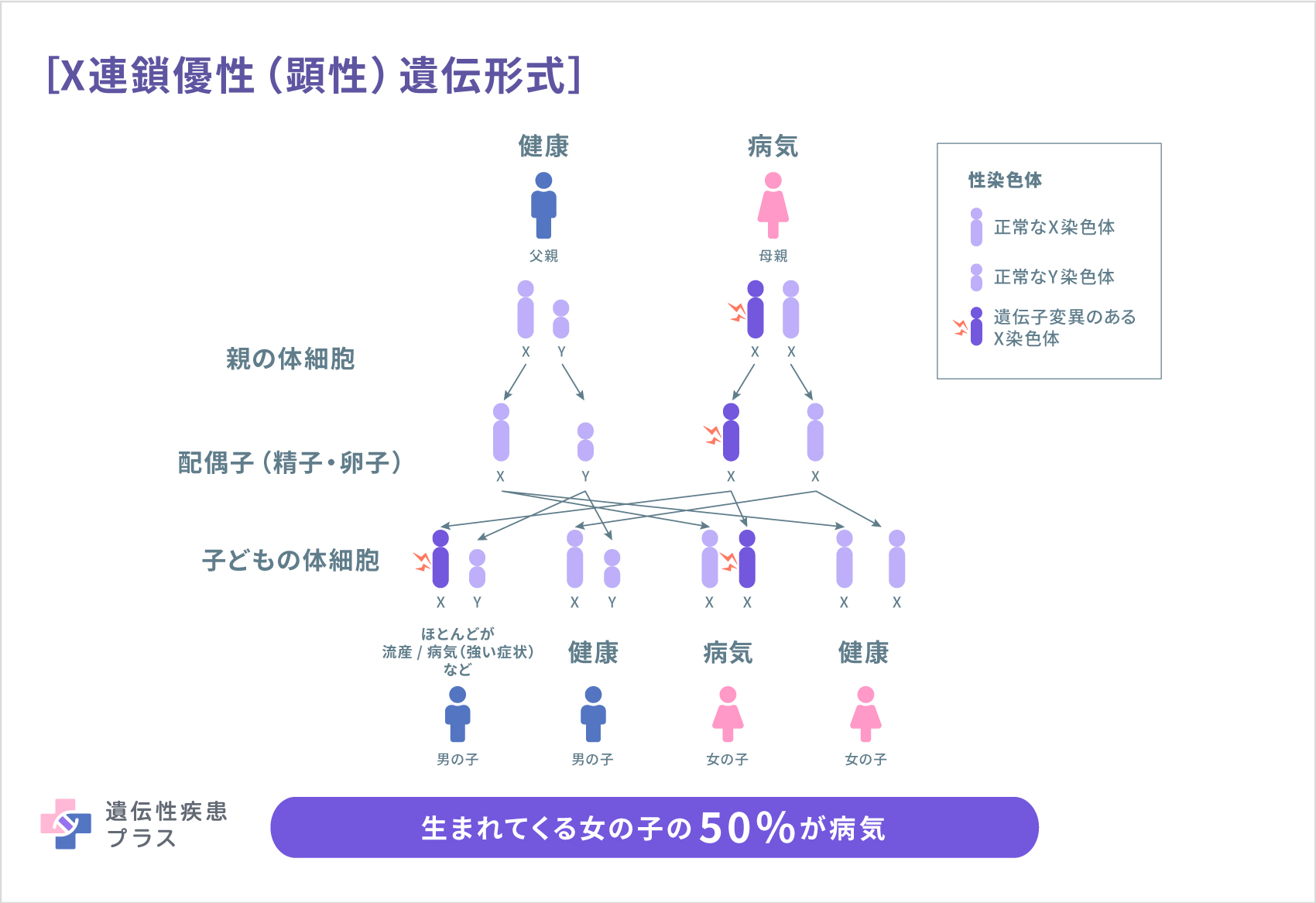
CDPX2はコレステロール合成の際に働くタンパク質であるEBPの産生に関与するEBP遺伝子の変異が関わっていると考えられています。EBPタンパク質は、3β-ヒドロキシステロイド Δ8,Δ7-イソメラーゼと呼ばれる酵素で、コレステロール生成の最終段階に重要な役割を持っています。コレステロールは、細胞膜を構成する成分であり、ホルモンなどの生成にも必要となります。そのため、正常な胚の発育に必須で、胎児期にも出生後にも重要な機能を果たします。EBP遺伝子の変異により、3β-ヒドロキシステロイド-Δ8,Δ7-イソメラーゼの活性が低下し、細胞が十分なコレステロールを生成できなくなるだけでなく、この酵素の不足によって、コレステロールが生成される前の状態の化合物が異常に体内に蓄積されます。コレステロール値の低下と他の物質の蓄積が組み合わさることで、体の多くの部分で成長や発達が妨げられる可能性があります。しかし、このコレステロール生成の障害がどのようにCDPX2のそれぞれの症状につながるのかははっきりわかっていません。
CDPX2はX連鎖性遺伝形式で遺伝します。母親がEBP遺伝子の変異を有する場合、子どもに変異のある遺伝子が受け継がれる確率は50%です。CDPX2では男児は致死的ですが、X染色体が2つある女児の場合にはその重症度はさまざまです。変異したEBP遺伝子を受け継いだ男児は妊娠早期に死亡するリスクが高く、CDPX2には男性の患者さんも報告されていますが、95%は女性です。
CHILD症候群の原因はNSDHL遺伝子の変異によると考えられています。この遺伝子も、EBP遺伝子と同様にコレステロールの生成に関与する酵素を作るための設計図となります。CHILD症候群はX連鎖性優性(顕性)遺伝形式で遺伝します。母親がNSDHL遺伝子の変異や欠損を持つ場合、子どもにその遺伝子が受け継がれる確率は50%です。
Keutel症候群の原因は異所性の石灰化(骨以外の組織にカルシウムが沈着すること)を抑制するMGP遺伝子の変異、Greenberg骨異形成症の原因はコレステロール生合成に関与するLBR遺伝子の変異であると知られています。これらはいずれも、常染色体劣性(潜性)遺伝形式で遺伝します。常染色体劣性(潜性)遺伝形式は両親がともに関連する遺伝子の片方に変異を持つ(保因者)場合、子どもは4分の1の確率で発症します。また、2分の1の確率で保因者となり、4分の1の確率でこの遺伝子の変異を持たずに生まれます。

脛骨・中手骨型点状軟骨異形成症は、原因となる遺伝子は不明です。常染色体優性(顕性)遺伝形式で遺伝するとされますが詳細はわかっていません。常染色体優性(顕性)遺伝形式は、親が病気の場合、病気が子どもに引き継がれる確率は50%です。

Astley-Kendall骨異形成症についても原因となる遺伝子はわかっていません。常染色体劣性(潜性)遺伝形式で遺伝すると考えられています。
どのように診断されるの?
小児慢性特定疾病情報センターの「点状軟骨異形成症(ペルオキシソーム病を除く。)」の診断基準は以下のようになっています。点状軟骨異形成症は症状が多彩で、幼児期までの点状軟骨の存在のみが共通項であるため、広義の点状軟骨異形成に関する診断基準が用いられています。
A.症状
1.鼻根部平坦と低い鼻
2.低身長
3.短い四肢
4.四肢骨の変形
B.検査所見
乳幼児期の骨、軟部の点状石灰化像
C.遺伝学的検査等
なし
D.鑑別診断
除外診断:近位肢型点状軟骨異形成症はペルオキシゾーム病であり除外
以下の疾患を鑑別:
1.骨形成不全症
2.軟骨無形性症
E-1. 確実例
Bがあり、Aのうち3項目以上を満たし、Dの鑑別すべき疾患を除外したもの
E-2. 疑い例
Bがあり、Aのうち2項目以上を満たし、Dの鑑別すべき疾患を除外したもの
Bを認めるもの
どのような治療が行われるの?
点状軟骨異形成症の根治的治療は確立されていないため、各症状に対する対症療法が行われます。頸椎椎体の骨化遅延、後弯変形、脊柱管狭窄、頸髄症、変形性関節症、四肢非対称などに対して、脊椎手術、関節手術、骨延長術などが行われます。米国のワシントン大学を中心としたスタッフが運営している遺伝性疾患情報サイト「GeneReviews」によると、CDPX1の呼吸困難に対しては鼻ステントや酸素投与、顎の形態異常に対しては必要に応じて再建術、不安定な頸椎に対しては頸椎固定術、頸椎閉塞に対しては減圧術、発達遅滞に対しては、理学療法、作業療法、言語療法などの治療が示されています。CDPX2に対する治療としては、骨格異常に対する治療、呼吸器合併症に対する治療、皮膚病変に対する皮膚軟化剤および角質溶解剤による治療、白内障に対する外科的治療、聴覚障害に対する耳鼻科医による標準的治療、水腎症に対する治療などが示されています。
どこで検査や治療が受けられるの?
日本で点状軟骨異形成症の診療を行っていることを公開している、主な施設は以下です。
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
難病の患者さん・ご家族、支えるさまざまな立場の方々とのネットワークづくりを行っている団体は、以下です。
参考サイト
- 難病情報センター
- 小児慢性特定疾病情報センター 点状軟骨異形成症(ペルオキシソーム病を除く。)
- MedlinePlus
- GeneReviews
- KEGG DISEASE:CHILD 症候群
- Online Mendelian Inheritance in Man(R) (OMIM(R))
- orphanet
- NORD
- Maceda EBG, et al., Novel NSDHL gene variant for congenital hemidysplasia with ichthyosiform erythroderma and limb defects (CHILD) syndrome., BMJ Case Rep.
- Lubala TK, et al., Greenberg Skeletal Dysplasia: first reported case in the Democratic Republic of Congo., Pan Afr Med J.





