



どのような病気?
コフィン・シリス症候群は、さまざまな程度の知的障害、座ったり歩いたり、しゃべったりなどの発達の遅れ、成長障害(低身長、体重増加の遅れなど)、「粗」と表現される細かい構造がはっきりとしない顔立ち、手足の「小指」の爪や指先の骨が無い/小さい、栄養をうまく摂取できない、呼吸器感染症を起こしやすい、などの症状がみられる、先天性の病気です。目、脳、心臓、腎臓に異常が存在する場合もあります。コフィン・シリス症候群の人に見られる顔の特徴はさまざまで、コフィン・シリス症候群に典型的な顔立ちというものはありません。
| コフィン・シリス症候群でみられる症状 |
|---|
99~80%でみられる症状 歯列の異常、小指の最も外側の骨がない/小さい(形成不全)、小指の爪がない/小さい(形成不全)、粗な顔立ち(顔の細かい構造がはっきりとしない)、乳児期の摂食困難、全身が毛深い、発達の遅れ、知的障害、長いまつ毛、小頭症、筋緊張が弱い、手の指の先端の骨が短い、低身長、髪の成長が遅い、眉が太い、下唇が厚い、鼻梁(びりょう)が広い |
79~30%でみられる症状 心血管系の形態異常、小脳の形成不全、膝蓋骨がない/小さい、停留精巣、脳の奇形(ダンディーウォーカー奇形)、鼻が低い/へこんでいる、肘の脱臼、難聴、出生前(子宮内胎児)発育不全、関節が可動域を超えて動く、眼振、呼吸器感染症を繰り返す、脊柱側弯症、発作、斜視、口が大きい |
29~5%でみられる症状 鎖骨の異常、股関節の骨の異常、椎間板の異常、足の小指の骨がない/小さい、手相が横一本、白内障、口蓋裂、先天性横隔膜ヘルニア、コクサバルガ(股関節の変形)、大理石様皮膚、腎臓の位置が異常、内眥贅皮(ないしぜいひ、蒙古ひだ)、水腎症、脳梁の形成不全、足の小指の爪が小さい、脊柱後湾症、涙液分泌異常、脳梁の部分欠損症、眼瞼下垂、腎臓がない/小さい、人中が短い、二分脊椎症 |
4~1%でみられる症状 強迫性行動 |
割合は示されていないがみられる症状 耳の形の異常、攻撃的な行動、爪がない、子宮がない、乱視、心房中隔欠損症、自閉症様行動、鼻先が広い、後鼻孔閉鎖(鼻の奥が塞がっている)、歯が生えるのが遅い、骨の成熟の遅れ、橈骨頭の脱臼、下向きの眼瞼裂、十二指腸潰瘍、顔面多毛症、胃潰瘍、血管腫、高い口蓋、甲高い声、嗄声、尿道下裂、眼球近接、鼠径ヘルニア、腸回転異常症、腸重積症、関節弛緩、耳の位置が低い、腰仙部多毛、近視、動脈管開存症、後ろに傾いた耳、子供の頃の成長の遅れ、先天性のいぼ、指の腹が目立つ、指の関節が目立つ、腎臓が小さい、仙骨部皮膚のへこみ、重度の表現性言語遅延、あごが短い、手足の小指の骨が短い、胸骨が短い、頭髪がまばら、ファロー四徴症(心臓の異常)、上唇が薄い、臍ヘルニア、心室中隔欠損症、視力障害 |
コフィン・シリス症候群は、まれな病気で、日本の患者さんは100人未満とされています。これまでの報告によると、男性よりも女性で頻繁に診断されています。
コフィン・シリス症候群は、国の指定難病対象疾患(指定難病185)、および、小児慢性特定疾病の対象疾患です。
何の遺伝子が原因となるの?
コフィン・シリス症候群の原因となる遺伝子は複数あり、今のところ、変異が見つかっている遺伝子は、SMARCB1、SMARCE1、SMARCA4、ARID1A、ARID1B、PHF6、SOX11、DPF2の8つです。最も多くみられるのは、ARID1B遺伝子の変異です。
これらの遺伝子は、「SWI/SNFタンパク質複合体」と呼ばれる、いくつかのタンパク質の集合体の構成要素(サブユニット)を作る設計図となっています。SWI/SNFタンパク質複合体は、「クロマチンリモデリング」と呼ばれる、細胞核の中で起こる重要な働きに関与しています。遺伝子の本体であるDNAは、クロマチンという構造を取ることで、染色体にきれいに詰め込まれています。この、クロマチン構造が、ゆるんだり、逆に緊密になったりするのが「クロマチンリモデリング」です。
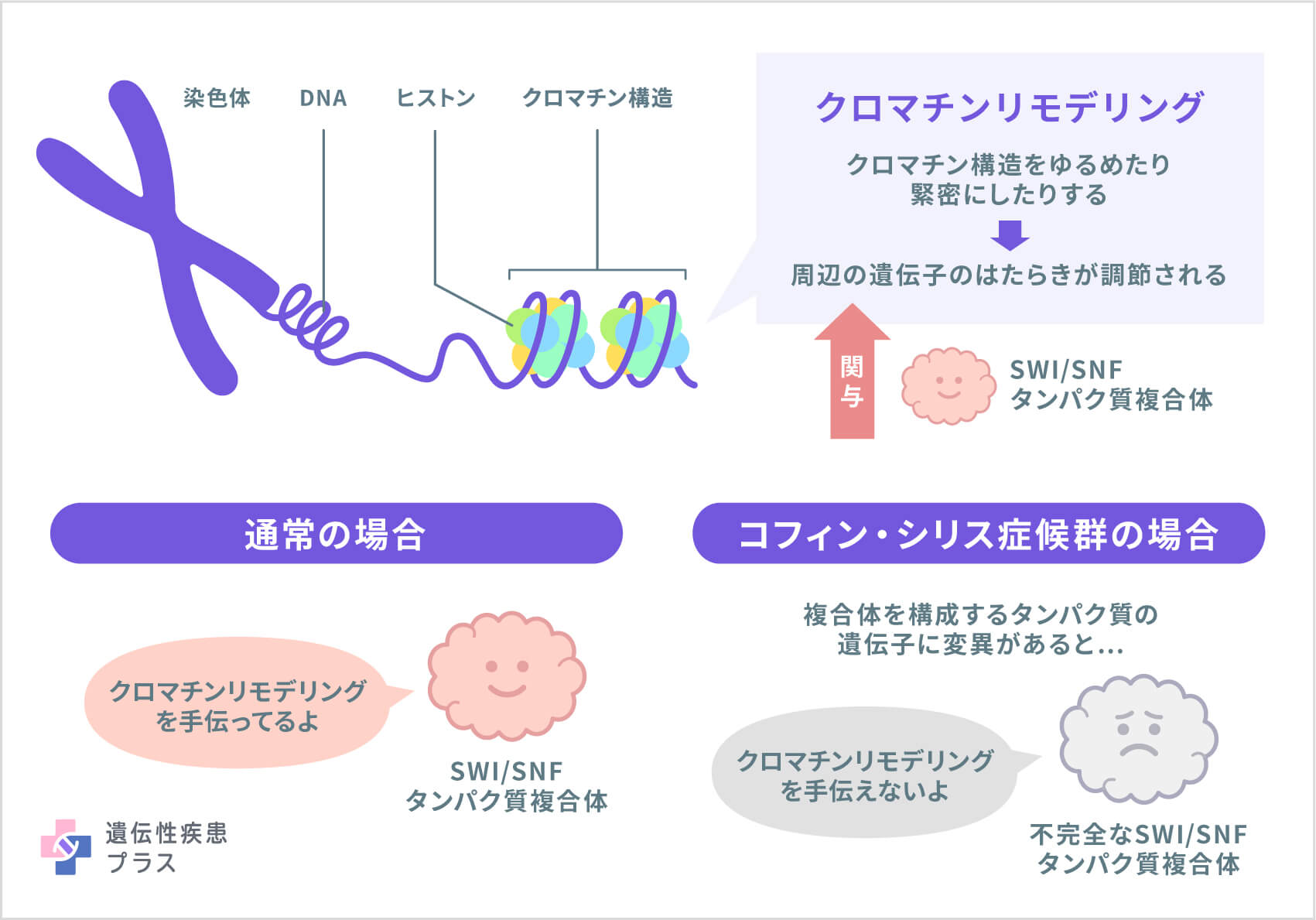
クロマチンリモデリングは、その構造変化が起きた周辺の遺伝子の働きを調節(活性化/抑制)するため、SWI/SNFタンパク質複合体は、損傷したDNAの修復、DNAの複製、細胞の成長、分裂、分化など、多くの重要な機能に関わっています。
| 遺伝子 | 染色体位置 | 作られるタンパク質の主な働き | 遺伝形式 |
| SMARCB1 | 22q11.23 | SWI/SNFタンパク質複合体のサブユニット | 常染色体優性(顕性) |
| SMARCE1 | 17q21.2 | SWI/SNFタンパク質複合体のサブユニット | 常染色体優性(顕性) |
| SMARCA4 | 19p13.2 | SWI/SNFタンパク質複合体のサブユニット | 常染色体優性(顕性) |
| ARID1A | 1p36.11 | SWI/SNFタンパク質複合体のサブユニット | 常染色体優性(顕性) |
| ARID1B | 6q25.3 | SWI/SNFタンパク質複合体のサブユニット | 常染色体優性(顕性) |
| DPF2 | 11q13.1 | SWI/SNFタンパク質複合体のサブユニット | 常染色体優性(顕性) |
| SOX11 | 2p25.2 | 遺伝子の転写に関与(転写因子) | 常染色体優性(顕性) |
| PHF6 | Xq26.2 | 遺伝子の転写に関与 | X連鎖劣性(潜性) |
これらの遺伝子の変異が、コフィン・シリス症候群の多様な症状に、どのように関わっているのかは、まだ研究が進められている段階で、完全には解明されていません。
コフィン・シリス症候群は、ほとんどが、親には遺伝子変異がなく、子どもで初めて発症する「孤発例」です。家族やきょうだいに同じ病気が見られる場合もありますが、その場合は、ほとんどが常染色体優性(顕性)遺伝形式で遺伝します。

どのように診断されるの?
コフィン・シリス症候群には、医師がコフィン・シリス症候群と診断するための「診断基準」があります。したがって、病院へ行き、必要な問診や検査を受けた後、主治医の先生がそれらの結果を診断基準に照らし合わせ、結果的にコフィン・シリス症候群かそうでないかの診断をすることになります。
具体的には、遺伝子検査で、これまでに見つかっている原因遺伝子のいずれかに変異が認められた場合、コフィン・シリス症候群と診断が確定します。
これらの遺伝子に変異が見つからない場合、乳・幼児期から「小指の爪と先端の骨が不完全/無い」「発達遅滞、知的障害」「コフィン・シリス症候群でみられる顔立ちの特徴」が全て認められると、コフィン・シリス症候群と臨床診断されます。
どのような治療が行われるの?
今のところ、コフィン・シリス症候群を根本的に、つまり、遺伝子から治すような治療法は見つかっていません。そのため、それぞれの症状に合わせた対症療法が行われます。例えば、てんかんに対しては必要に応じて薬物療法、心疾患に対しては必要に応じて手術や薬物療法が行われます。
起きやすい症状に対しては、早期から検診をして適切な治療を行っていきます。また、発達の遅れに対しては、療育的支援(理学療法、作業療法、言語療法)を行っていきます。
世界的にも、まだコフィン・シリス症候群に特化した治療法の開発は行われていませんが、それに向けての第一歩は、コフィン・シリス症候群の発症メカニズムを十分に解明することです。そうした研究は、世界各国で行われており、米国国立衛生研究所(NIH)の米国国立医学図書館(NLM)が運営する、世界の医学論文データベース”PubMed”で「Coffin-Siris syndrome」と検索すると、2023年5月現在、276件の研究論文がヒットします。
どこで検査や治療が受けられるの?
日本でコフィン・シリス症候群の診療を行っていることを公開している、主な施設は以下です。
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
難病の患者さん・ご家族、支えるさまざまな立場の方々とのネットワークづくりを行っている団体は、以下です。
米国のコフィン・シリス症候群の患者支援団体(財団)で、ホームページを公開しているところは、以下です。
参考サイト
- 難病情報センター
- 小児慢性特定疾病情報センター コフィン・シリス(Coffin-Siris)症候群
- MedlinePlus
- Genetic and Rare Diseases Information Center
- Online Mendelian Inheritance in Man(R) (OMIM(R))






