



どのような病気?
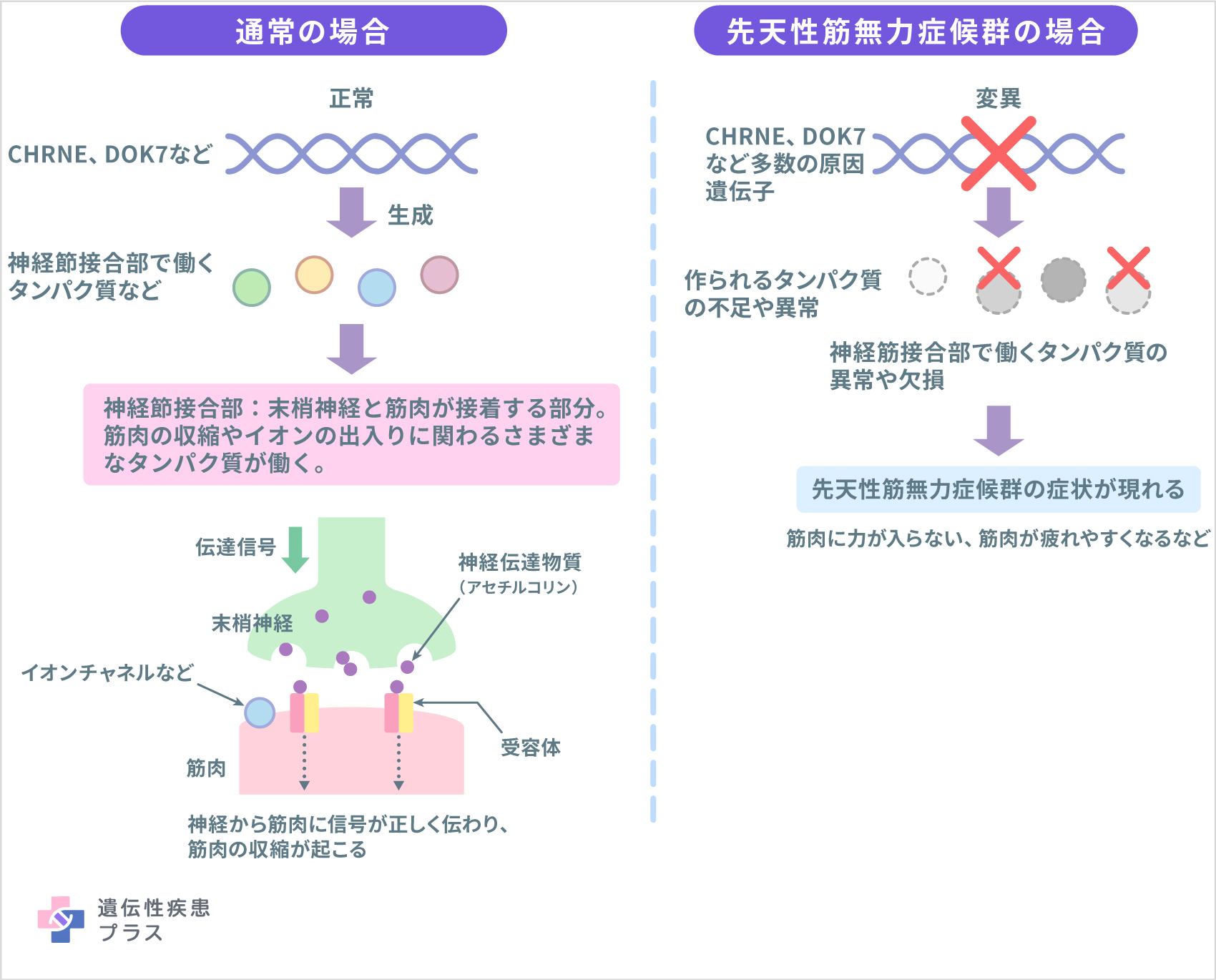
先天性筋無力症候群は体のさまざまな部位の筋肉に力が入らなかったり、筋肉が疲れやすくなる疾患です。ヒトが体を動かすためには筋肉の収縮が必要です。脳からの信号が中枢神経から末梢神経を経て、筋肉に伝わることで筋肉が収縮します。この末梢神経と筋肉の接合部(神経筋接合部)では、末梢神経からアセチルコリンという神経伝達物質が分泌され、それが筋肉側にあるアセチルコリン受容体に結合することで筋肉に信号が伝わります。また、筋肉の収縮には筋肉の細胞内外へのイオン(荷電した原子)の移動が関与し、イオンが移動するための通路が開く必要があります。神経筋接合部には筋肉の収縮やイオンの出入りに関わるさまざまなタンパク質が存在します。これらのタンパク質の欠損や機能障害が生じることで先天性筋無力症候群が発症します。
先天性筋無力症候群は主に、神経伝達物質であるアセチルコリンの受容体が欠損する「終板アセチルコリン受容体欠損症」、アセチルコリン受容体のイオンチャネル(チャンネル)の開口時間が延長する「スローチャンネル症候群」、イオンチャネルの開口時間が短縮する「ファーストチャンネル症候群」、骨格筋ナトリウムチャネルの開口に異常を生じる「ナトリウムチャンネル筋無力症」、アセチルコリン分解酵素が欠損する「終板アセチルコリンエステラーゼ欠損症」、神経終末のアセチルコリン再合成酵素が欠損する「発作性無呼吸を伴う先天性筋無力症」の6種類に分けられ、その他に、エスコバー症候群、致死的多発性翼状片症候群、胎児無動変形シークエンス、先天性ランバートイートン筋無力症候群なども、先天性筋無力症候群に含まれます。
筋力低下は、新生児期の泣き声が小さい、母乳を吸う能力が弱いなどの軽度のものから、呼吸困難で人工呼吸器が必要となるなどの重度のものまでさまざまです。多くの場合、出生直後に見られるこれらの症状はいったん軽快し、幼児~小児期に再度、持続的な筋力低下や筋無力症状が見られます。
先天性筋無力症候群で見られる症状 |
|---|
高頻度に見られる症状 嚥下障害(食べ物を飲み込みにくい状態)、易疲労性・虚弱性、摂食障害、前頭筋筋力低下(前頭筋は頭部前面にある筋肉)、筋力低下による呼吸不全、首の筋力低下、哺乳力低下、近位筋力低下(近位筋は体幹に近い筋)、眼瞼下垂、発作性無呼吸 |
よく見られる症状 病気・疲労・ストレスなどによって誘発される無呼吸、先天性多発性関節拘縮症、運動失調、球まひ(延髄または脳幹周辺の神経障害によって生じるまひ)、中枢性睡眠時無呼吸、呼吸困難、チアノーゼ(低酸素血症による皮膚や粘膜の変色)、胎動低下(妊娠中の胎児の動きの減少)、歩行障害、呼吸窮迫、全身筋力低下、知的障害、筋繊維萎縮、鼻逆流(飲食物が鼻へ逆流すること)、鼻声、眼筋まひ、反復する呼吸器感染症、脊椎奇形 |
しばしば見られる症状 反射消失、末梢性筋委縮症、下肢遠位部筋力低下(遠位部は体幹から遠い部分)、発声障害、高口蓋(上あごの前歯の裏側のくぼみが深いこと)、筋緊張低下、後弯症(脊柱が後ろに曲がっている状態)、肢帯筋力低下(肢体筋は対になった肢を脊柱に結合する部分の筋肉で肩と腰にある)、長い顔、運動発達遅滞、狭い下あご、凹足(足のアーチと甲が高い)、頭部の位置の維持困難、てんかん発作、脊髄性固縮、吸気性喘鳴(吸気時の喘鳴)、つま先歩行、動揺歩行(よたよた歩き)、弱い泣き声 |
まれに見られる症状 先天性股関節脱臼、複視(物が二つに見えること)、内斜視、胃食道逆流、反射減退、関節弛緩症、低い位置の耳、後方にある下あご、末梢性運動神経障害、眼振(意思と関係なく眼が動くこと)、閉塞性睡眠時無呼吸(気道の閉塞による睡眠時無呼吸)、鳩胸、羊水過多、呼吸停止、感音性聴覚障害(内耳や中枢神経系の障害による聴覚障害)、凝視 |
先天性筋無力症候群の有病率は不明で、世界の患者数は少なくとも600人とされています。また、国内の患者数は約20人とされています。先天性筋無力症候群は診断が困難で国内外ともに診断されていない患者さんが多いと考えられています。
先天性筋無力症候群は、指定難病対象疾病(指定難病12)です。
何の遺伝子が原因となるの?
神経筋接合部で筋肉の収縮に関与するタンパク質の産生に関わる遺伝子の変異よって、十分な量のタンパク質を作ることができない、あるいは正常に機能するタンパク質が産生されないことが先天性筋無力症候群の原因です。原因となる遺伝子として難病情報センターでは19種類(CHRNE、RAPSN、COLQ、DOK7、CHAT、CHRNA1、CHRNB1、CHRND、AGRN、LRP4、MUSK、LAMB2、SCN4A、GFPT1、DPAGT1、ALG2、ALG14、PLEC、PREPL)が伝えられています。また、難治性疾患等政策研究事業の希少難治性筋疾患に関する調査研究班が作成した先天性筋無力症候群診療の手引きによれば、2022年12月の時点で、(AGRN、CHD8、CHRNG、COL13A1、GMPPB、LAMA5、MYO9A、PURA、RPH3A、SLC18A3、SLC25A1、SLC5A7、SNAP25、SYT2、TOR1AIP1、 UNC13A、VAMP1)を加えた35種類の遺伝子が報告されていると記載されています。
中でもCHRNE遺伝子の変異が全体の50%程度を占めるとされています。その他に割合が高い遺伝子はRAPSN(15~20%)、COLQ(10~15%)、DOK7(10~15%)、CHAT(4~5%)とされています。
先天性筋無力症候群の遺伝形式は、その病型によって異なります。スローチャンネル症候群は常染色体優性(顕性)遺伝形式で遺伝します。その他の終板アセチルコリン受容体欠損症、ファーストチャンネル症候群、ナトリウムチャンネル筋無力症、終板アセチルコリンエステラーゼ欠損症、発作性無呼吸を伴う先天性筋無力症はいずれも常染色体劣性(潜性)遺伝形式で遺伝します。常染色体優性(顕性)遺伝形式は、両親のどちらかがこの病気だった場合、子どもは50%の確率で発症します。常染色体劣性(潜性)遺伝形式は、両親がともに遺伝子の片方に変異を持つ(保因者)場合、子どもは4分の1の確率で発症します。また、2分の1の確率で保因者となり、4分の1の確率でこの遺伝子の変異を持たずに生まれます。


どのように診断されるの?
国内の難病情報センターのサイトには以下の診断基準が示されています。
- 2歳以下で発症の骨格筋易疲労性、骨格筋低形成、および反復神経刺激による複合筋活動電位の異常減衰により先天性筋無力症候群を疑い、遺伝子異常により診断する。
- 確定診断には19種類(CHRNA1、CHRNB1、CHRND、CHRNE、COLQ、AGRN、LRP4、MUSK、LAMB2、RAPSN、DOK7、CHAT、SCN4A、GFPT1、DPAGT1、ALG2、ALG14、PLEC、PREPL)の遺伝子を対象とした遺伝子診断を行う。
- 臨床補助診断としては、重症筋無力症において認められる抗体(抗アセチルコリン受容体抗体・抗MuSK抗体・抗LRP4抗体)が陰性であることに加えて、反復神経刺激による異常な複合筋活動電位の減衰が必要条件である。
どのような治療が行われるの?
先天性筋無力症候群は、種類にもよりますが、有効な薬剤が存在するものがあります。終板アセチルコリン受容体欠損症やファーストチャンネル症候群に対しては、アセチルコリンの分解を抑制する抗コリンエステラーゼ剤やアセチルコリンの放出を促進する3,4-ジアミノピリジンが、治療薬として使用されます。終板アセチルコリンエステラーゼ欠損症とDok7筋無力症に対しては交感神経刺激剤のエフェドリンが使用されます。また、スローチャンネル症候群に対してナトリウムチャネルを抑制するキニジンや神経伝達物質に作用するフルオキセチンが使用されます。ナトリウムチャンネル筋無力症に対してはアセタゾラミドが使用されます。
先天性筋無力症候群は進行性ではありませんが症状は継続します。呼吸筋の筋力低下や易疲労性に伴う呼吸困難が見られることがあり、特に発作性無呼吸を伴う先天性筋無力症は、乳児突然死症候群の原因となることがあるので睡眠時の呼吸モニターが行われます。脊柱筋の脱力による脊柱側弯がある場合には、必要に応じて手術による矯正が行われます。
どこで検査や治療が受けられるの?
日本で先天性筋無力症候群の診療を行っていることを公開している、主な施設は以下です。
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
難病の患者さん・ご家族、支えるさまざまな立場の方々とのネットワークづくりを行っている団体は、以下です。
参考サイト
- 難病情報センター
- MedlinePlus
- Genetic and Rare Diseases Information Center
- 厚生労働科学研究費 難治性疾患等政策研究事業 希少難治性筋疾患に関する調査研究班、「先天性筋無力症候群診療の手引き(指定難病12)」






