



どのような病気?
先天性ミオパチーは、筋力低下や筋緊張低下のほか、呼吸障害、関節拘縮などさまざまな症状が見られる遺伝性疾患です。ミオは筋肉、パチーは病気を意味し、先天性とは、症状などが生まれつき見られる、ということを意味しています。
この病気は、生まれつき筋肉の組織の構造に異常が見られ、多くは生後間もなくから幼少期までの間において、上記のような筋力低下に関わる症状が見られる、歩行開始が遅れる、といった運動発達の遅れによって発見されます。
この病気で見られる症状や重症度は、この病気に含まれる複数の病型や遺伝子変異、また個人によっても異なりますが、主な症状として、顔面の筋肉を含む全身の筋緊張低下、高口蓋(上あごの前歯の裏側のくぼみが深いこと)、眼瞼(まぶた)が下がる、呼吸困難、哺乳や嚥下障害(飲み込みがうまくいかない)、発育・発達の遅れ、関節の拘縮、脊柱側弯症などが見られます。また、知的障害、てんかん、心臓の合併症(心筋症、不整脈など)が見られることもあります。
呼吸の症状としては、呼吸に関わる筋力が弱くなることで、肺での酸素の取り込みと二酸化炭素の排出に問題が生じる換気障害が見られることがあります。換気障害は多くの場合夜から始まり、ゆっくり進行するため、日中、息苦しいなど感じることはありませんが、起床時にぼんやりしたり頭痛がする、痰が出しにくいなどから自覚することがあります。哺乳や嚥下障害は、食欲の低下や体重の減少など、栄養や成長の面でも注意が必要となります。
進行や重症度のパターンもさまざまですが、大きく乳児重症型、良性先天型、成人発症型の3つに分けられます。最も重症な乳児重症型では、新生児期から命に関わるような強い呼吸障害や哺乳障害が見られ、生まれてすぐに酸素や人工呼吸器の使用が必要になったり、ミルクを飲む力が弱く経管での栄養投与が必要になるなどします。良性先天型では、乳児期からの筋緊張低下や、発育・発達の遅れが見られますが歩行することはでき、進行しないかもしくはゆっくり進行します。成人発症型は、大人になってから力が入りにくい、疲れやすいといった症状が急に見られるなどから発見される場合があります。
先天性ミオパチーは、筋肉の組織を一部採取して顕微鏡などで調べる「病理学的検査」での特徴をもとに、いくつかの病型に分類されます。病型には、「ネマリンミオパチー」「セントラルコア病」「ミニコア病」「ミオチュブラーミオパチー」「中心核ミオパチー」「先天性筋線維タイプ不均等症」「全タイプ1線維ミオパチー」などがあります。このほかにも病型は存在するほか、病理学的検査で特徴的な所見を示さないことから分類できない先天性ミオパチーも存在します。
先天性ミオパチーの病型と特徴
病型 | 病理学的検査で見られる特徴 | 症状や特徴 |
|---|---|---|
ネマリンミオパチー | ネマリン小体と呼ばれる棒状、もしくは糸状の構造が筋細胞の中に確認される。「ネマ」はギリシャ語で「糸」を意味する | ・全身の筋力が低下するが、特に顔、首、体幹、上腕、脚など体の中心に近い筋肉(近位筋)の筋肉に症状が見られる ・筋力低下は時間とともに悪化する可能性がある ・症状が現れる年齢と重症度によって重症先天型、アーミッシュ型、中等度先天型、典型的先天性、小児期発症型、成人発症型に区別される ・重症先天型は最も重症、幼少期に命に関わる症状が見られる ・成人発症型はタイプの中で最も軽症、20~50歳の間に筋力低下を発症 |
セントラルコア病 | ミトコンドリアの活性が見られないコアと呼ばれる部分が筋細胞の中心に見られる | ・筋力低下は、ほとんど気づかない程度から非常に重度までさまざま ・多くは、軽度の筋力低下が進行せず継続する ・筋力低下は、近位筋の中で特に肩、大腿部、股関節に影響する ・斜視、脊柱後側弯症、足の変形、股関節脱臼などが見られる場合がある ・手術などの処置中に使用される麻酔などに反応して頻脈、発熱(急激な上昇)、頻呼吸、筋肉硬直、筋線維の破壊、アシドーシスなどの重篤な症状が現れる「悪性高熱症」のリスクが高いとされる |
ミニコア病(マルチミニコア病) | 活性低下の範囲が小さなコアが筋細胞に多数見られる | ・古典型、手への影響を伴う進行型、関節拘縮を伴う出生前型、眼筋麻痺型に分けられ、最も多いのは古典型 ・古典型は、体幹と首の筋肉(体幹筋)に症状が見られることが多く、腕と脚の筋肉はそれほど影響を受けない ・眼筋麻痺型は、外眼筋麻痺が特徴で、異常な眼球運動や眼瞼下垂が生じることがある。近位筋の筋力低下も引き起こすことがある ・セントラルコア病と同様に悪性高熱症リスクが高いとされる |
中心核ミオパチー | 筋細胞の核の位置に異常が見られる。通常は周縁部にある核が、細胞の中心に位置している | ・重症度は同じ家族内でも異なる ・筋力低下の発症は出生後~成人初期までさまざま ・筋力低下は時間とともにゆっくりと悪化する ・筋萎縮と筋力低下が重症化すると、車椅子による介助が必要になる場合がある ・まれに、筋力低下が時間の経過とともに改善する場合が報告されている |
ミオチューブラーミオパチー | 筋線維が全体的に小型で未熟であり、胎児期の筋線維(ミオチューブ)に類似している。筋細胞の中央に大きな核が位置する | ・ほとんどの場合男性だけに発症 ・出生時から筋力低下と筋緊張低下が見られることが多い ・重度の呼吸障害が幼少期の生命予後に影響することが多い ・筋力低下により正常な骨の発達が阻害され、骨粗鬆症、側弯症、股や膝の関節拘縮につながることがある ・患者さんは、頭が大きい、顔が細長い、口蓋が高いなどの特徴が見られることがある ・耳や呼吸器の感染症、てんかん発作、肝疾患を繰り返すなども見られる。一部で、紫斑病性肝炎と呼ばれる重篤な肝疾患を発症することがある |
筋線維タイプ不均等症 | タイプ1筋線維(遅筋線維)がタイプ2筋線維(速筋線維)と比較して小さい | ・特に肩、上腕、股関節、大腿部の筋肉に筋力低下が見られる傾向 ・筋力低下は顔面筋や眼球運動を制御する筋肉にも影響を及ぼす可能性があり、眼筋麻痺や眼瞼下垂が見られる場合がある ・顔が長い、高口蓋、歯が密集してはえるなどの特徴が見られることが多い ・約30%で軽度から重度の呼吸障害が見られる ・嚥下障害も約30%に見られる ・まれに、拡張型心筋症が見られることがある ・重症度は個人により大きく異なる ・筋力低下は進行しないことが多く、場合によっては改善することもあるとされるが、まれに、徐々に筋力が低下し車いすが必要になることもある |
全タイプ1線維ミオパチー | タイプ2筋線維(速筋線維)が見られず、ほぼすべての筋線維がタイプ1筋線維(遅筋線維)である | ・まれな病型 ・乳児期の筋緊張低下、軽度の近位筋筋力低下、呼吸困難、高口蓋、頭蓋や顔立ちの特徴など、他の病型と同様とされる |
先天性ミオパチーの国内における正確な頻度と患者数はわかっていませんが1,000~3,000人の患者さんがいると推定されています。海外では、およそ1.7万出生に1人の割合で発症すると報告されており、病型ではネマリンミオパチーとセントラルコア病の頻度が高いとされています。
先天性ミオパチーは、指定難病対象疾病(指定難病111)、および「ミオチュブラーミオパチー」「先天性筋線維不均等症」「ネマリンミオパチー」「セントラルコア病」「マルチコア病」「ミニコア病」「その他、先天性ミオパチー」として、それぞれ小児慢性特定疾病の対象となっています。
何の遺伝子が原因となるの?
先天性ミオパチーでは、それぞれの病型で、複数の原因遺伝子が明らかになっています(下表参照)
ネマリンミオパチー
染色体領域 | 原因遺伝子 | 遺伝形式 |
|---|---|---|
1q42.13 | ACTA1 | 常染色体優性(顕性)または常染色体劣性(潜性)遺伝 |
2q23.3 | NEB | 常染色体優性(顕性)または常染色体劣性(潜性)遺伝 |
3p22.1 | KLHL40 | 常染色体劣性(潜性)遺伝 |
1q21.3 | TPM3 | 常染色体優性(顕性)または常染色体劣性(潜性)遺伝 |
9p13.3 | TPM2 | 常染色体優性(顕性)遺伝 |
19q13.2 | RYR1 | 常染色体劣性(潜性)遺伝 |
19q13.42 | TNNT1 | 常染色体劣性(潜性)遺伝 |
14q13.1 | CFL2 | 常染色体劣性(潜性)遺伝 |
15q22.31 | KBTBD13 | 常染色体優性(顕性)遺伝 |
3p14.1 | LMOD3 | 常染色体劣性(潜性)遺伝 |
10q21.3 | MYPN | 常染色体劣性(潜性)遺伝 |
2q31.1 | KLHL41 | 常染色体劣性(潜性)遺伝 |
セントラルコア病
染色体領域 | 原因遺伝子 | 遺伝形式 |
|---|---|---|
19q13.2 | RYR1 | 常染色体優性(顕性)または常染色体劣性(潜性)遺伝 |
ミニコア病
染色体領域 | 原因遺伝子 | 遺伝形式 |
|---|---|---|
1p36.11 | SELENON | 常染色体劣性(潜性)遺伝 |
19q13.2 | RYR1 | 常染色体劣性(潜性)遺伝 |
ミオチュブラーミオパチー
染色体領域 | 原因遺伝子 | 遺伝形式 |
|---|---|---|
Xq28 | MTM1 | X連鎖劣性(潜性)遺伝 |
中心核ミオパチー
染色体領域 | 原因遺伝子 | 遺伝形式 |
|---|---|---|
19p13.2 | DNM2 | 常染色体優性(顕性)遺伝 |
2q14.3 | BIN1 | 常染色体劣性(潜性)遺伝 |
19q13.2 | RYR1 | 常染色体劣性(潜性)遺伝 |
2q31.2 | TTN | 常染色体劣性(潜性)遺伝 |
2q35 | SPEG | 常染色体劣性(潜性)遺伝 |
16p13.3 | CCDC78 | 常染色体優性(顕性)遺伝 |
筋線維タイプ不均等症
染色体領域 | 原因遺伝子 | 遺伝形式 |
|---|---|---|
1q21.3 | TPM3 | 常染色体優性(顕性)または常染色体劣性(潜性)遺伝 |
19q13.2 | RYR1 | 常染色体劣性(潜性)遺伝 |
1q42.13 | ACTA1 | 常染色体優性(顕性)または常染色体劣性(潜性)遺伝 |
14q11.2 | MYH7 | 常染色体優性(顕性)遺伝 |
9p13.3 | TPM2 | 常染色体優性(顕性)遺伝 |
1p36.11 | SELENON | 常染色体劣性(潜性)遺伝 |
12q24.11 | MYL2 | 常染色体劣性(潜性)遺伝 |
全タイプ1線維ミオパチー
染色体領域 | 原因遺伝子 | 遺伝形式 |
|---|---|---|
19q13.2 | RYR1 | 常染色体優性(顕性)遺伝 |
先天性ミオパチーの原因遺伝子の多くは、骨格筋のタンパク質や骨格筋の機能に関連しています。しかし、それぞれの原因遺伝子と症状の関連などの詳細については、未解明な点も多く残っています。病理学的検査で典型的な病型と診断されても、上記の遺伝子変異が認められないこともあり、先天性ミオパチー全体で少なくとも半数以上において原因となる遺伝子変異がわかっていません。
また、遺伝子によっては、同じ遺伝子変異であっても病理学的検査からは異なる病型となることがあり、遺伝子変異だけでは病型を確定できないことがあります。また、病型や遺伝子変異は、必ずしも重症度と関連している訳ではありません。
先天性ミオパチーでは、遺伝子の新たな変異によって家族歴のない人に発生することも多いとされます。
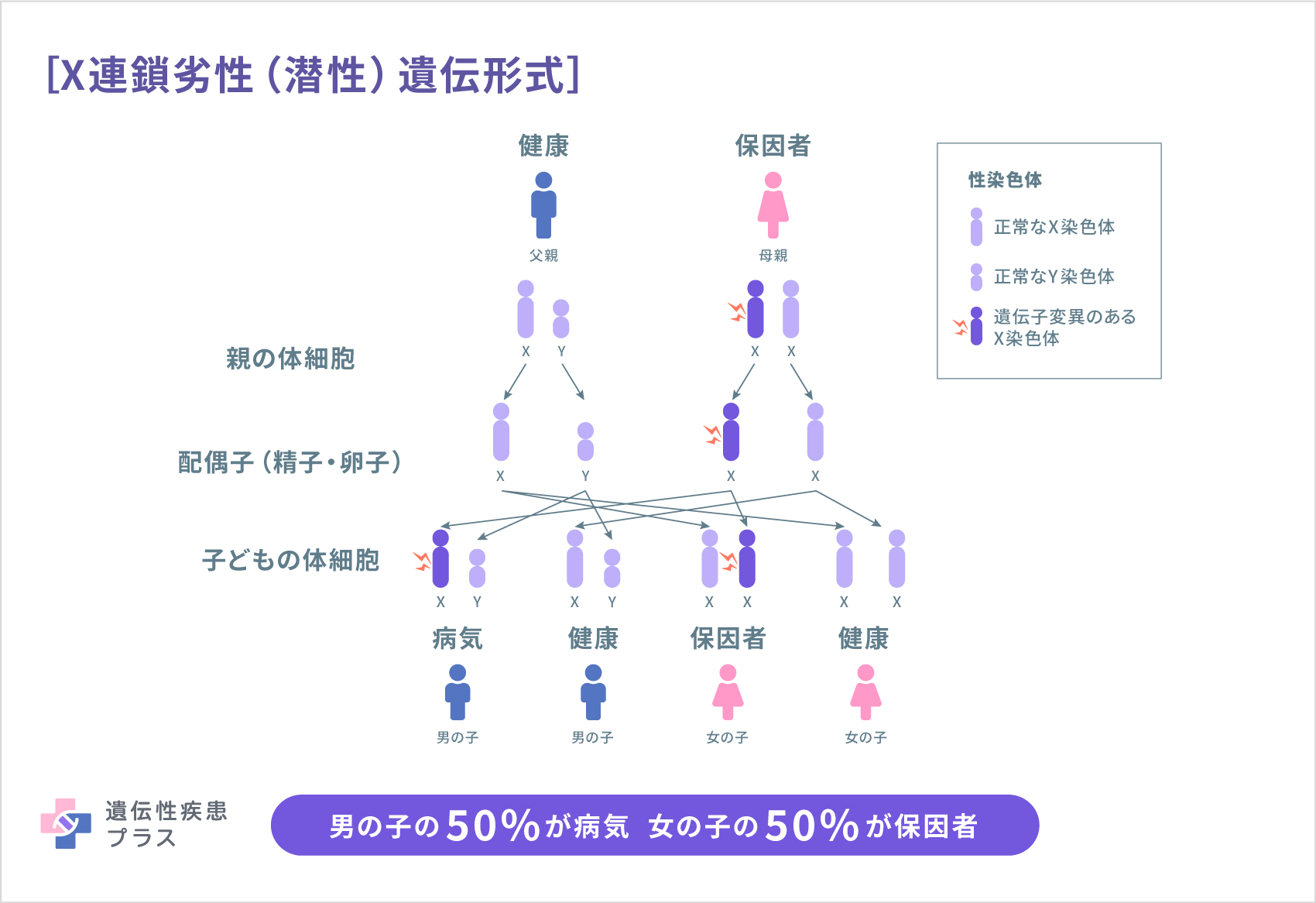
遺伝する場合、先天性ミオパチーの遺伝形式は、上記の表のように、多くは常染色体優性(顕性)遺伝形式、または常染色体劣性(潜性)遺伝形式となり、ミオチュブラーミオパチーのみX連鎖劣性(潜性)遺伝形式となります。
常染色体優性(顕性)遺伝形式では、父母のどちらかがこの病気である場合に子どもがこの病気を発症する可能性は50%です。

常染色体劣性(潜性)遺伝形式では、父母から受け継いだ両方の遺伝子に変異があることで発症します。両親はこの病気の保因者で、病気は発症しません。

X連鎖劣性(潜性)遺伝形式では、病気が遺伝する確率が男女で異なります。男性(XY)はX染色体が1つであり、その遺伝子に変異があると発症します。X染色体を2つ持つ女性(XX)は、1つの遺伝子に変異があっても、男性の場合よりも軽症であるか、全く症状が現れないこともあります。
どのように診断されるの?
先天性ミオパチーの診断では、血液検査、筋電図検査、骨格筋画像検査などが行われ、検査結果から先天性ミオパチーの可能性があると考えられる場合には、さらに精密検査が行われます。診断の確定には、筋組織を採取して、顕微鏡で筋組織の構造を確認する病理学的検査(筋生検)や遺伝学的検査が必要になります。
難病情報センターによれば、先天性ミオパチーの診断基準は以下のように記載されています。
(1)臨床症状
1.筋力低下
新生児期:自発運動の低下
乳幼児:運動発達の遅れ
学童~成人:徒手筋力テストで複数筋が4以下
2.筋緊張低下
3.腱反射の低下または消失
(2)検査所見
1.筋生検で特徴的な病理所見を認める
2.先天性ミオパチーで既報の原因遺伝子に変異が同定されている
(家族で同症を呈し遺伝子が確定している例も可)
(3)その他の所見
1.骨格筋画像(CTまたはMRI)で萎縮・異常信号輝度を認める
2.呼吸機能障害があり人工呼吸器を要する
3.経鼻胃管または胃瘻による経管栄養を要する
4.側弯または関節拘縮を認める
5.顔面筋罹患または高口蓋、眼瞼下垂、外眼筋麻痺を認める
6.家族歴あり
<診断のカテゴリー>
1)または2)の場合にこの病気であると診断されます
1)(1)臨床症状のいずれかを満たし、かつ(2)検査所見のいずれかの検査で所見を認める
2)(1)臨床症状のいずれかを満たし、(2)検査所見は未実施または所見なしだが、(3)その他の所見を3つ以上認める
※2)の場合は、20歳以下で診断し、中枢神経病変の否定、骨格筋画像・針筋電図・遺伝子検査で筋炎や神経原性疾患の除外、染色体異常の否定、CK値低下~軽度上昇が必須
どのような治療が行われるの?
先天性ミオパチーを根本的に治療する方法はまだ確立されていません。そのため、症状に合わせた対症療法が中心となります。それぞれの症状に対し、いかに早く気が付き治療を開始するかがQOLを高めるために重要となります。
筋力低下や側弯症、関節の拘縮などには、リハビリテーション、装具、手術療法などが行われます。先天性ミオパチーを含む8つの神経・筋疾患に対し、歩行リハビリテーションとしてHAL医療用下肢タイプによる歩行運動処置が2016年から保険適用されています。
呼吸障害には、人工呼吸器、痰を排出しやすくしたりする呼吸リハビリテーションなども行われることがあります。
心臓の症状に対しては、薬が処方されることもあります。その他には体重の維持や栄養管理なども行われます。
ミオチュブラーミオパチーでは、遺伝子治療の臨床研究が進められています。
どこで検査や治療が受けられるの?
日本で先天性ミオパチーの診療を行っていることを公開している、主な施設は以下です。
- 東京大学医学部附属病院 小児科
- 順天堂大学医学部附属順天堂医院 脳神経内科筋疾患部門
- 東京女子医科大学小児科
- 東京都立小児総合医療センター 神経内科
- 国立成育医療研究センター 神経内科
- 大阪母子医療センター
全国遺伝子医療部門連絡会議 登録機関遺伝子医療体制検索・提供システムに登録されている神経・筋疾患ミオパチーを診療している医療機関は、こちらからご確認頂けます
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
先天性ミオパチーの患者会で、ホームページを公開しているところは、以下です。
参考サイト
- 難病情報センター
- 小児慢性特定疾病情報センター ネマリンミオパチー
- 小児慢性特定疾病情報センター セントラルコア病
- 小児慢性特定疾病情報センター マルチコア病
- 小児慢性特定疾病情報センター ミオチュブラーミオパチー
- 小児慢性特定疾病情報センター 先天性筋線維不均等症
- 小児慢性特定疾病情報センター 56から61までに掲げるもののほか、先天性ミオパチー
- MedlinePlus
- Online Mendelian Inheritance in Man(R) (OMIM(R))
- KEGG DISEASE:先天性ミオパチー
- 国立医薬品食品衛生研究所、「承認申請中・第3相臨床試験段階にある主な遺伝子治療製品 (2025年4月4日更新)」
- Sato I, et al., Congenital neuromuscular disease with uniform type 1 fiber and RYR1 mutation., Neurology.
- Lawal TA, et al., Ryanodine Receptor 1-Related Myopathies: Diagnostic and Therapeutic Approaches., Neurotherapeutics.






