



どのような病気?
コルネリア・デランゲ症候群は、特徴的な顔立ち、成長の遅れ、知的障害を伴う全般性発達遅滞、手足の異常など体の多くの部位に影響が現れる遺伝性疾患です。この病気は、1933年にオランダの小児科医であるコルネリア・カタリーナ・デ・ランゲによって報告されたことから名づけられました。
コルネリア・デランゲ症候群によく見られる症状として、特徴的な顔立ちが挙げられます。具体的には、左右がつながった眉毛、カールした長いまつげ、低い位置にある耳、小さくて上向きの鼻、隙間の多い歯などが特徴で、顔以外に小さい頭(小頭症)や体毛が多いといった外観的特徴もみられます。成長の遅れは多くの場合、出生前(妊娠後期ごろ)からみられ、生後6か月までに低身長が際立ってきます。ほとんどの人に発達遅滞が見られますが、知的障害は軽度から重度と幅広く、個人差があります。手足については、手足の形成不全(主に腕、手、指の異常)、側弯症、合趾症(足の指がつながっている状態)などがみられます。
消化器症状の中では、胃食道逆流症が多く、バレット食道(食道の粘膜が胃の粘膜のように変化して、食道がんになりやすい)などにつながることもあります。吐き気、下痢、便秘などの一般的な症状の他、幽門狭窄症(胃から十二指腸への出口が狭くなる)、腸回転異常、横隔膜ヘルニアなども報告されています。これらの消化器症状が、哺乳障害や摂食不良につながることで栄養がうまくとれず、さらに2次的な成長障害を引き起こす可能性もあります。聴覚障害は、感音性難聴(音を感じる神経に原因)や伝音性難聴(鼓膜や内耳に原因)が子どもの約80%以上にみられますが、どちらの難聴も成長とともに改善されると言われています。眼科系では眼瞼下垂、斜視、眼振、近視、白内障、緑内障などがあり、何らかの視覚障害が全体の50%程度にみられます。
その他の症状として、泌尿生殖器系では停留精巣、性器発育不全、尿道下裂、腎臓の構造的異常などがみられます。約30%に心血管系の症状があり、心房中隔欠損、心室中隔欠損などの他に、肺動脈狭窄、ファロー四徴症(心室中隔欠損、大動脈騎乗、肺動脈狭窄、右室肥大の4つを合併した状態)の報告もあります。行動上では、コミュニケーションや社会との関わりにおいて、自閉症とよく似た問題や症状が現れる以外に、注意欠如・多動性障害(ADHD)、うつ、不安障害、強迫性障害などもみられます。
コルネリア・デランゲ症候群は、軽度から重度まで幅広い程度の症状が見られる病気であることが知られています。それは、70年以上前に典型的(古典的)なコルネリア・デランゲ症候群として、症状から定義されていたこの病気が、近年になって遺伝的な原因が少しずつ明らかになり、症状はそれほど顕著ではないものの、遺伝子の変異にこの病気の原因と一致した特徴を持つ人が、軽度のコルネリア・デランゲ症候群として認識される例が増えてきたことが背景としてあるためです。
古典的なコルネリア・デランゲ症候群の場合、症状は重度であり、顕著な発育不全(成長曲線が同年齢の5パーセンタイル未満)、重度から最重度の知的障害、前腕や指の欠損などがみられることが多いとされます。軽度の場合は、顔立ちはこの病気の特徴と一致していることが多いものの、成長障害、認知機能、足や腕への症状は深刻ではないとされます。
| コルネリア・デランゲ症候群で見られる症状 |
|---|
高頻度に見られる症状 短頭症、小頭症、低い前頭部毛髪線(生え際と眉間の間の距離が短い)、眉毛癒合(左右の眉毛がつながっている)、太い眉毛、弓形の眉毛、長いまつげ、カール状のまつげ、外耳道閉鎖症(耳の穴が閉鎖もしくは非常に狭い)、上向きの鼻孔、低い鼻梁、小さい鼻、長い人中(唇上部の溝)、高口蓋、薄い紅唇、口角下垂、歯萌出遅延(歯が生えるのが遅いこと)、隙間の多い歯、小顎症、短い首、低い後頭部毛髪線、小さい手、親指の形態異常、短い第1中手骨(親指の手首から指まで伸びる骨)、小肢症(通常よりも小さいまたは短い手足)、短い足、つま先の合趾症、低身長、異常に低い声、骨格成熟遅延、胃食道逆流症、全身性の多毛、筋緊張亢進(腕や脚が硬くなり、動きにくくなるなど)、知的障害(中度~重度)、摂食困難 |
良く見られる症状 眼瞼下垂、眼瞼炎(まぶたの炎症)、小角膜、眼球癆(眼球の萎縮により機能しない状態)、低い位置で後方に傾いた耳、肘関節脱臼、前椀骨(橈尺骨)の癒合、手掌単一屈曲線(手のひらに一直線に伸びる深いしわ)、第5指の湾曲(斜指症)、乳頭低形成、大陰唇低形成、陰茎低形成、停留精巣、尿道下裂、多嚢胞性腎異形成、膀胱尿管逆流症、大理石様皮膚(網の目の赤紫色の皮斑)、関節硬直、近視、伝音難聴、感音難聴、(胎児期の)子宮内発育遅延、未熟児、重度出生後発育遅延、哺乳障害、成長障害、神経学的発語障害、不安症、注意欠如・多動性障害(ADHD)、強迫性障害、睡眠障害 |
しばしば見られる症状 大耳症、口蓋裂、尺骨(前腕の小指側の骨)形態異常、欠指(乏指)症、漏斗胸(胸骨の一部が陥没している)、股関節脱臼、股関節形成不全、湾足、末梢神経障害、筋緊張低下、脳室肥大、大脳皮質の萎縮、小脳低形成、心血管系形態異常、心房中隔欠損、心室中隔欠損、幽門狭窄症、腸回転異常、腸捻転、子宮形態異常、眼振、斜視、白内障、緑内障、後鼻孔閉鎖(鼻の奥が塞がっている)、先天性横隔膜ヘルニア、自閉症、出生前運動異常、思春期遅発症、原発性無月経、腎不全、てんかん、体幹の肥満、脊柱側弯症 |
この病気は、軽度な場合にこの病気だと気が付かれない場合があり、正確な有病率ははっきりしていませんが、およそ出生1万~3万人に1人の頻度で発症すると推定されています。
コルネリア・デランゲ症候群は小児慢性特定疾病に指定されています。また、国の指定難病対象疾患である先天異常症候群(指定難病310)に該当する場合もあります。
何の遺伝子が原因となるの?
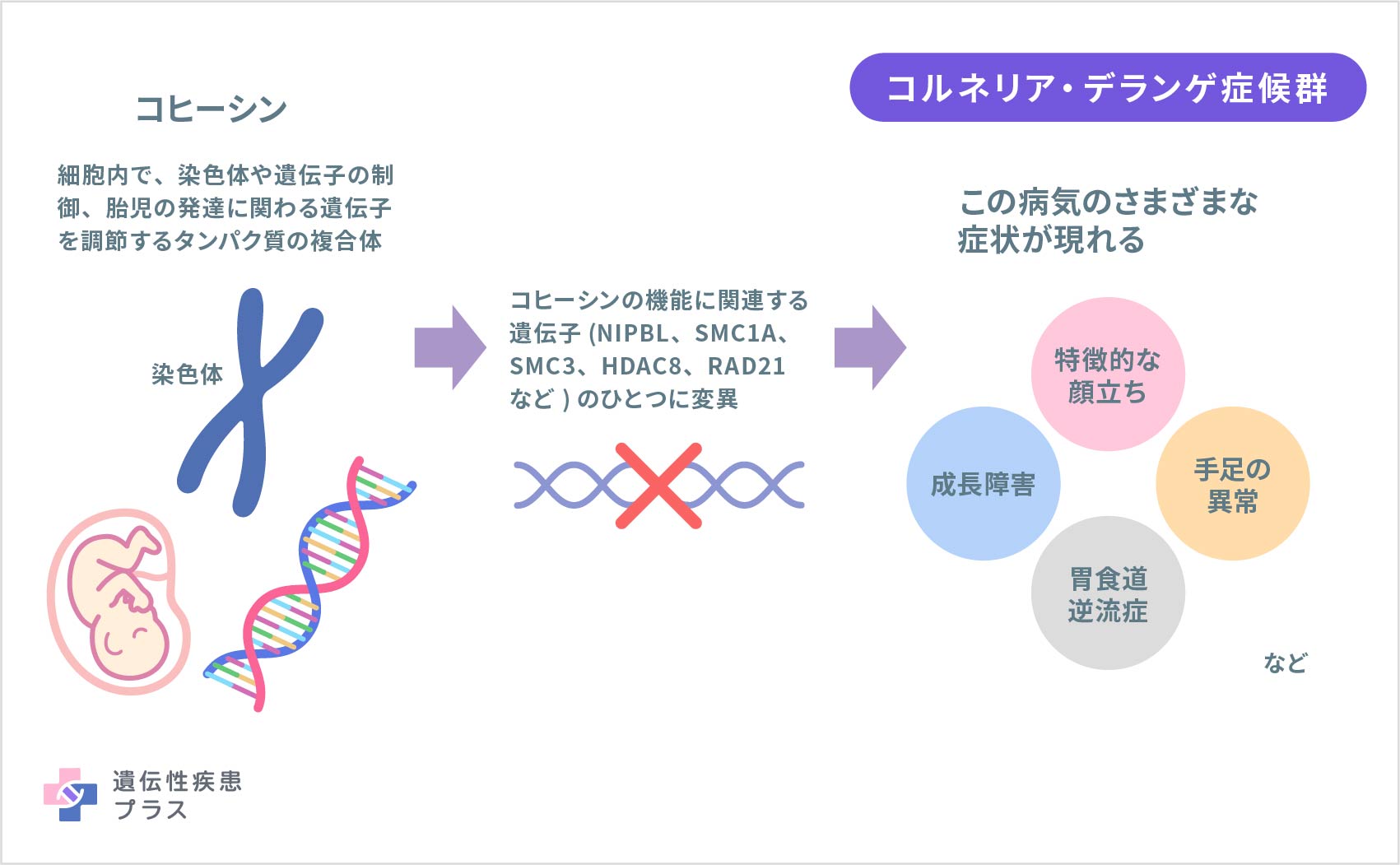
コルネリア・デランゲ症候群は、複数の遺伝子のうちの1つに変異が起こることが原因として知られています。これまでに、この病気の原因となりうる遺伝子として、「NIPBL(染色体位置:5p13.2)」「SMC1A(Xp11.22)」「SMC3(10q25.2)」「HDAC8(Xq13.1)」「RAD21(8q24.11)」「BRD4(19p13.12)」「ANKRD11(16q24.3)」などの遺伝子が報告されてきました。
これらの遺伝子は、コヒーシンの構造や機能に関わるタンパク質の設計図であることが知られています。コヒーシンは、細胞の中で、染色体構造の制御、細胞の遺伝情報の安定化、DNA損傷の修復など、染色体や遺伝子の基本的な働きにおける重要な調節に関わるタンパク質の複合体です。またコヒーシンは、顔、手足、他の体の部分を含む胎児の頃の発達に関わる遺伝子を調節する機能もあるとされます。原因とされる遺伝子の1つが変異することにより、コヒーシンの機能が妨げられ、発達に関連する遺伝子の制御が失われることでコルネリア・デランゲ症候群が発症すると考えられています。
これまでのところ、この病気の60%以上においてNIPBL遺伝子の変異が原因であり、それ以外のSMC1A、SMC3、HDAC8、RAD21、BRD4、ANKRD11遺伝子などの変異が原因となる例は合わせても10%前後であると考えられます。一方で、コルネリア・デランゲ症候群のおよそ15%においては遺伝的な原因がわかっていません。
NIPBL遺伝子の変異は多くの場合、古典的と呼ばれる重度の症状を示し、NIPBL遺伝子変異が原因となる場合と比較すると、SMC1A、RAD21、SMC3遺伝子の変異は軽症であることが多いとされます。またHDAC8遺伝子の変異は、乳児期の頭部にある大泉門の閉鎖が遅れる、目の間隔の広さ、歯の異常など、やや異なる症状を引き起こすとされます。知的障害はNIPBLとHDAC8遺伝子の変異の場合には重症化する可能性があります。
コルネリア・デランゲ症候群の発症は、多くの場合親からの遺伝ではなく孤発例であり、新規に生じた遺伝子の変異が原因で起こります。両親のどちらかがこの病気を持つ場合、変異している遺伝子によって子どもへの遺伝形式が異なります。この病気の原因がNIPBL、RAD21、SMC3遺伝子の場合には、常染色体優性(顕性)遺伝形式で遺伝します。これらの遺伝子の変異を有する場合、子どもが病気を受け継ぐ確率は50%です。

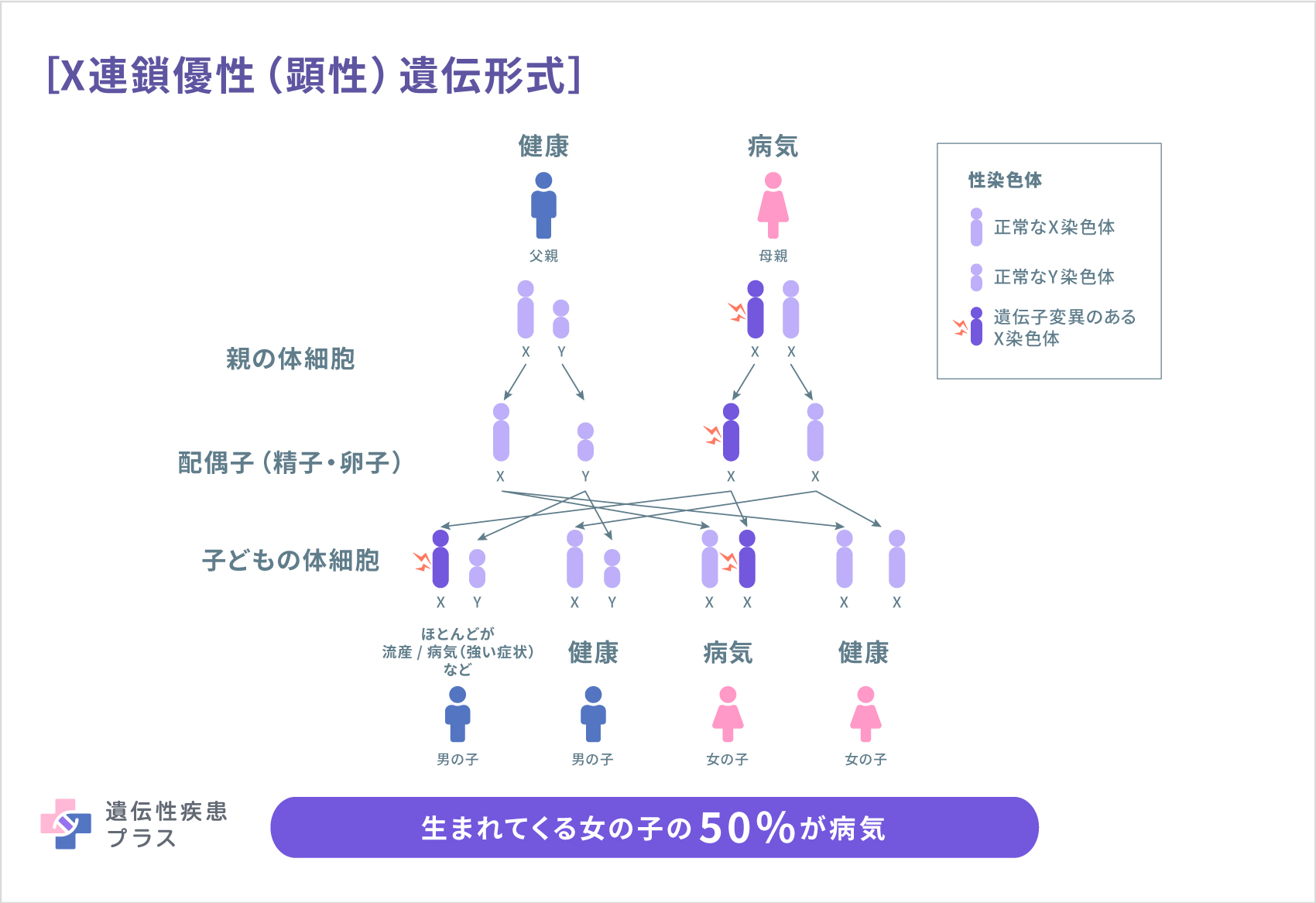
病気の原因がHDAC8またはSMC1A遺伝子の変異である場合には、これらの遺伝子はX染色体と呼ばれる性染色体に位置しているため、X連鎖優性(顕性)遺伝形式で遺伝します。この遺伝形式では、病気が遺伝する確率は男女で異なります。ヒトは、性染色体と呼ばれるX染色体とY染色体の2種類の染色体を1対2本持っています。男性はXとYの組み合わせで、女性はXとXの組み合わせになります。男性はY染色体を息子に、X染色体を娘に受け継がせるため、息子には引き継がれませんが、娘には必ずそのX染色体が受け継がれます。女性の場合は、50%の確率でその原因遺伝子が存在するX染色体を娘または息子に受け継がせることになります。また、X染色体の遺伝子変異の場合、男性はX染色体が1本のみですが、女性はX染色体を2本持っているため、1本が異常でももう1本の遺伝子が正常に働くことで機能を補完し、多くの場合男性に比べて軽症となります。
どのように診断されるの?
この病気の診断は、
- 眉毛癒合(左右の眉毛がつながっている)
- 知的障害
- 成長障害(身長ないし体重が3パーセンタイル未満)
上記の3つの症状があり、さらに遺伝学的診断により、NIPBL、SMC1A、RAD21、SCC1、SMC3、HDAC8遺伝子等の原因遺伝子に変異が認められればコルネリア・デランゲ症候群の診断が確定します。
また、遺伝学的検査で変異が認められない場合は、上記の3つの症状に加えて以下の3つの症状があればコルネリア・デランゲ症候群と診断されます。
- 長い人中または薄い上口唇
- 長いまつげ
- 小肢症、第5指短小または乏指症
どのような治療が行われるの?
コルネリア・デランゲ症候群に対する根治的な治療法はなく、さまざまな症状に対する対症療法が中心となります。多様な症状がみられることから、その治療に際しては主治医のほか、神経科、耳鼻科、消化器科、眼科、心臓外科、泌尿器科などの多科連携による治療が行われます。
難聴がある場合には、早期に補聴器の使用を開始し、サイン言語や身振り・手振りを取り入れるなどコミュニケーションを補うことに重点が置かれます。発達遅延や知的障害に対しては、理学療法、言語聴覚療法、作業療法なども考慮されます。摂食不良、消化器合併症、停留精巣、骨格異常などで外科的な治療が必要な場合には手術も選択肢となります。その他、必要な場合には、定期的な消化管検査、成長、精神運動機能の発達、視覚、聴覚、心機能、腎機能の検査と観察が行われます。
どこで検査や治療が受けられるの?
日本でコルネリア・デランゲ症候群の診療を行っていることを公開している、主な施設は以下です。
- 埼玉県立小児医療センター遺伝科
- 国立成育医療研究センター遺伝診療科
- 東京医療センター臨床遺伝センター
- 東京女子医科大学病院ゲノム診療科
- 愛知県医療療育総合センター中央病院
- 大阪母子医療センター遺伝診療科
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
コルネリア・デランゲ症候群の患者会で、ホームページを公開しているところは、以下です。
参考サイト
- 難病情報センター
- 小児慢性特定疾病情報センター コルネリア・デランゲ(Cornelia de Lange)症候群
- MedlinePlus
- Genetic and Rare Diseases Information Center
- Online Mendelian Inheritance in Man(R) (OMIM(R))
- GeneReviews
- StatPearls







