



どのような病気?
エーラス・ダンロス症候群は、皮膚、関節、血管、その他多くの全身の臓器や組織を支える「結合組織」の脆弱性(もろさ)を特徴とする遺伝性疾患です。その原因と症状から、1998年に6つの主な病型(古典型、関節型、血管型、後側弯型、多発関節弛緩型、皮膚弛緩型)とその他の病型に分類されましたが、その後、新たな病型が次々に見つかっており、2017年に発表された新たな国際分類には、13の病型が示されています。これらの病型の特徴は、重複する部分があります。その他、この13の病型に含まれない、まだ十分に特徴付けられていない非常にまれなケースもあります。
エーラス・ダンロス症候群で見られる症状は、人によってさまざまですが、主な症状は以下です。
| EDSの13の病型と主な症状 |
|---|
古典型EDS(Classical EDS) 皮膚が伸びやすく非常に弾力性がある(過伸展性)、皮膚が滑らかで脆い(裂けやすい、傷つきやすい、内出血しやすい、特徴的な傷跡(萎縮性瘢痕)が広範囲に残るなど)、関節の可動域が通常の範囲を超えて大きく脱臼しやすい、モルスクム様偽腫瘍(肘などにできる石灰化血腫)、スフェロイド(前腕やすねにできる脂肪含有嚢胞)、筋緊張低下、運動能力の発達の遅れ、組織のもろさによるヘルニア、直腸脱、心血管異常(僧帽弁逸脱症や大動脈根拡張症など)。女性は、前期破水のリスクあり。 |
関節可動亢進型EDS(Hypermobile EDS) 大きな関節(肘、ひざ)・小さな関節(指・つま先)どちらも可動域が通常の範囲を超えて大きい、関節の脱臼・亜脱臼を繰り返す、皮膚が柔らかく滑らかでやや伸展性があり青あざができやすい、筋肉と骨の慢性痛、早期発症型変形性関節症、骨粗しょう症、運動障害、胃腸の問題(腹部膨満、吐き気、嘔吐、胸焼け、便秘、裂孔ヘルニア、など)、自律神経系の機能障害、心血管異常(僧帽弁逸脱症や大動脈根拡張症など)など。女性では、骨盤臓器脱、月経困難症、性交疼痛症などのリスクの増加、前期破水や早産(4時間未満)などの妊娠合併症リスクの増加など。 |
血管型EDS(Vascular EDS) 薄くて半透明の傷つきやすい皮膚、動脈・腸・子宮など特定の臓器が壊れやすく破裂しやすい、特徴的な顔立ち(薄い唇、小さなあご、細い鼻、大きな目)、アクロジェリア(手足の皮膚の早期老化)、小さな関節(指・つま先)の可動域が通常の範囲を超えて大きい、静脈瘤の早期発症、気胸、あざができやすい、関節の脱臼・亜脱臼、先天性股関節脱臼、先天性内反足、歯肉退縮など。 |
後側弯型EDS(Kyphoscoliosis EDS) もろくて傷つきやすくあざができやすい過伸展性の皮膚、関節の可動域が通常の範囲を超えて大きく脱臼・亜脱臼を繰り返す、出生時の重度の筋緊張低下、出生時または生後1年以内に認められる進行性の後側弯症、白目(強膜)の部分が脆弱、傷の治り方の異常、マルファン症候群様の体型(長くて細い指、異常に長い手足、漏斗胸または鳩胸)、破裂しやすく壊れやすい動脈、運動能力の発達の遅れ、異常に小さい角膜、低骨密度、先天性内反足、心血管異常(僧帽弁逸脱症や大動脈根拡張症など)など。 |
多発関節弛緩型EDS(Arthrochalasia EDS) 重度の関節過可動性、関節の脱臼・亜脱臼を繰り返す、先天性股関節脱臼、もろくて傷つきやすくあざができやすい過伸展性の皮膚、筋緊張低下、運動能力の発達の遅れ、後側弯症、低骨密度、特徴的な顔立ち(目の間隔が広い、内眼角贅皮、泉門が大きい、小さなあご)など。 |
皮膚弛緩型EDS(Dermatosparaxis EDS) 柔らかくてたるんだ非常に傷つきやすい皮膚、重度のあざと傷跡(瘢痕)、特に顔の皮膚がたるんでいる、ヘルニア、低身長、泉門の閉鎖の遅れ、短い指、特徴的な顔立ち(ふくらんだまぶた、白目が青い、内眼角贅皮、下向きの眼瞼裂、小さなあご)、膀胱や横隔膜の破裂、関節の過可動性(軽度~重度)など。 |
脆弱角膜症候群(Brittle Cornea Syndrome、BCS) 薄い角膜、早期発症の進行性球状角膜、青い強膜(白目の部分)を特徴とする。 |
類古典型(Classical-like EDS、clEDS) ベルベットのような肌質で萎縮性瘢痕はなく過伸展性の皮膚、全身性関節過可動性(主に肩や足首の脱臼を繰り返す場合もある)、傷つきやすい皮膚、自発性の内出血を特徴とする。 |
脊椎異形成型EDS(Spondylodysplastic EDS、spEDS) 低身長(小児期に進行)、筋緊張低下(重度の先天性から軽度の後天性までさまざま)、手足の反り返りを特徴とする。 |
筋拘縮型EDS(Musculocontractural EDS、mcEDS) 先天性多発性拘縮(内転拘縮、屈曲拘縮、内反足など)、特徴的な頭蓋顔面の特徴(出生時または乳児期早期)、皮膚の特徴(過伸展性、あざができやすい、萎縮性瘢痕を伴う皮膚の脆弱性、てのひらのしわの増加など)を特徴とする。 |
ミオパチー型EDS(Myopathic EDS、mEDS) 先天性の筋緊張低下および/または筋萎縮がみられ年齢とともに改善する。その他、近位の関節拘縮(膝、股関節、肘の関節)および遠位の関節(足首、手首、足、手の関節)の過可動性を特徴とする。 |
歯周型EDS(Periodontal EDS、pEDS) 早期発症(小児期または思春期)の重度で難治性の歯周炎、付着歯肉の欠如、前歯部プラーク、両親のどちらかが同じ臨床的診断基準を満たしていることを特徴とする。 |
心臓弁型EDS(Cardiac-valvular EDS、cvEDS) 重度の進行性心臓弁膜症(大動脈弁、僧帽弁)、皮膚の異常(過伸展性、萎縮性瘢痕、皮膚が薄い、あざができやすい)、関節の過可動性(全身または小さな関節に限定)を特徴とする。 |
症状は、小児期・若年成人期から生涯続き、生活の質(QOL)に影響があります。重症度は、軽度の関節の緩みから命に関わる合併症までさまざまで、こうした重症度や経過は、合併症と治療の状況により異なります。一般的に、血管型EDSが最も重症な病型と考えられています。女性の場合、妊娠分娩時の合併症も多く、妊娠管理も重要とされています。
EDSの全病型を含む世界的な発症頻度は約5,000人に1人と推定されており、日本に患者さんは約2万人いると推定されています。最も一般的な病型は、古典型と関節可動亢進型で、古典型は2万~4万人に1人、関節可動亢進型は5,000~2万人に1人の頻度で発症すると考えられています。その他の病型はまれで、医学文献での症例報告も多くありません。
EDSは、国の指定難病対象疾患(指定難病168)、および、小児慢性特定疾病の対象疾患です。
何の遺伝子が原因となるの?
EDSの原因遺伝子は、複数見つかっています。その中には、いくつかの異なるタイプのタンパク質「コラーゲン断片」の設計図となる遺伝子や、コラーゲンの成熟に必要な酵素の遺伝子が含まれています。例えば、COL1A1、COL1A2、COL3A1、COL5A1、COL5A2などの遺伝子からコラーゲン断片が作られ、それらが集まって成熟したコラーゲン分子を形成することで、体全体の結合組織が強度をもって成り立ちます。ADAMTS2、FKBP14、PLOD1、TNXBなどの遺伝子は、コラーゲンを処理するタンパク質、折り畳むタンパク質、または相互作用するタンパク質などの設計図となっています。こうした遺伝子に変異が生じると、コラーゲンが適切に構成されなくなり、皮膚や骨をはじめ、全身の結合組織が弱くなり、エーラス・ダンロス症候群となります。しかし、それぞれの遺伝子変異がどのような仕組みで複数の合併症につながるのか、治療につながる詳細なメカニズムはまだ解明されていません。
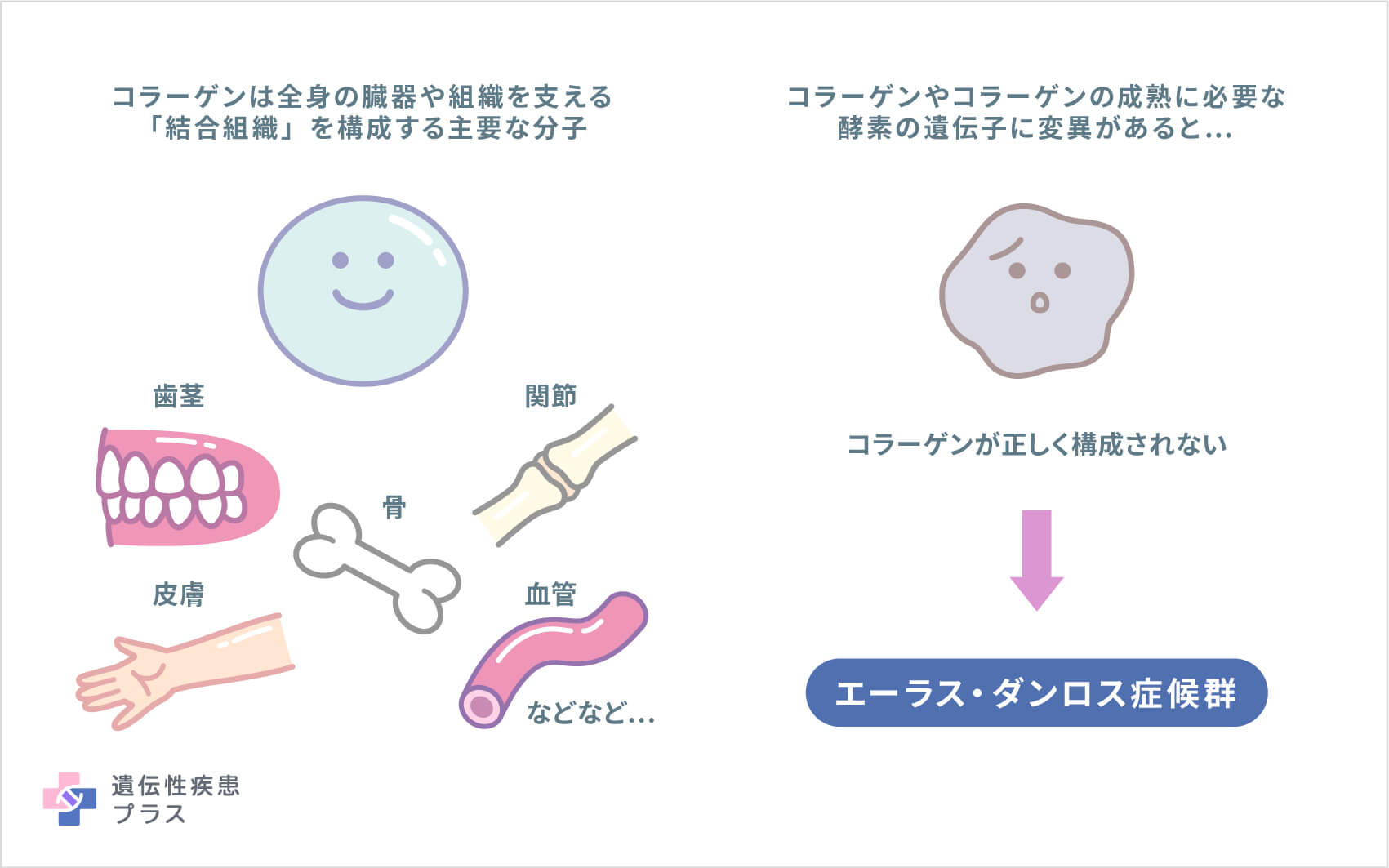
最近、エーラス・ダンロス症候群に関連する新たな遺伝子として、コラーゲンとは関連がなさそうな遺伝子もいくつか報告されてきています。それらの遺伝子の多くは、変異がどのような仕組みで病気の発症につながるのか、まだ明らかにされていません。
2017年の新たな病型分類の発表論文に示されている、各病型と原因遺伝子は下表の通りです。(染色体位置はOMIMを参照して追記。)
| 病型(略称) | 遺伝形式 | 原因遺伝子(染色体位置) | 作られるタンパク質 |
|---|---|---|---|
| 古典型EDS(cEDS) | 常染色体優性(顕性)遺伝 | 多くがCOL5A1(9q34.3)、COL5A2(2q32.2) まれにCOL1A1(17q21.33) | COL5A1とCOL5A2はⅤ型コラーゲン、COL1A1はI型コラーゲン |
| 類古典型EDS(clEDS) | 常染色体劣性(潜性)遺伝 | TNXB(6p21.33-p21.32) | テネイシンXB |
| 心臓弁型EDS(cvEDS) | 常染色体劣性(潜性)遺伝 | COL1A2(7q21.3) | Ⅰ型コラーゲン |
| 血管型EDS(vEDS) | 常染色体優性(顕性)遺伝 | 多くがCOL3A1(2q32.2) まれにCOL1A1(17q21.33) | COL3A1はIII型コラーゲン、COL1A1はI型コラーゲン |
| 関節可動亢進型EDS(hEDS) | 常染色体優性(顕性)遺伝 | 不明 | 不明 |
| 多発関節弛緩型EDS(aEDS) | 常染色体優性(顕性)遺伝 | COL1A1(17q21.33)、COL1A2(7q21.3) | I型コラーゲン |
| 皮膚弛緩型EDS(dEDS) | 常染色体劣性(潜性)遺伝 | ADAMTS2(5q35.3) | ADAMTS-2(酵素) |
| 後側弯型EDS(kEDS) | 常染色体劣性(潜性)遺伝 | PLOD1(1p36.22) FKBP14(7p14.3) | LH1(酵素) FKBP14※ |
| 脆弱角膜症候群(BCS) | 常染色体劣性(潜性)遺伝 | ZNF469(16q24.2) PRDM5(4q27) | ZNF469 PRDM5 |
| 脊椎異形成型EDS(spEDS) | 常染色体劣性(潜性)遺伝 | B4GALT7(5q35.3) B4GALT6(18q12.1) SLC39A13(11p11.2) | β4GalT7(酵素) β4GalT6(酵素) ZIP13 |
| 筋拘縮型EDS(mcEDS) | 常染色体劣性(潜性)遺伝 | CHST14(15q15.1) DSE(6q22.1) | D4ST1(酵素) DSE(酵素) |
| ミオパチー型EDS(mEDS) | 常染色体優性(顕性)遺伝 または常染色体劣性(潜性)遺伝 | COL12A1(6q13-q14) | ⅩⅡ型コラーゲン |
| 歯周型EDS(pEDS) | 常染色体優性(顕性)遺伝 | C1R(12p13.31) C1S(12p13.31) | C1r(補体成分) C1s(補体成分) |
※論文にはFKBP22と記されているが、OMIM上でFKBP14であることを確認。
EDSは、常染色体優性(顕性)遺伝形式または常染色体劣性(潜性)遺伝形式のいずれかの形式で遺伝します。親から変異を受け継いで病気を発症するケース以外にも、血のつながった家系内にEDSの病歴がなく、両親ともにEDSではない場合でも、生まれてくる過程で原因遺伝子に変異が起きて、EDSになるケースがあります。


どのように診断されるの?
皮膚症状(過伸展性や脆弱性)、関節症状(過可動性や弛緩性)、家族歴からEDSの疑いとなった場合、臨床所見からどの病型に当てはまるか医師が調べ、さらにほかの疾患でないことを確かめたうえで(鑑別診断)、EDSと診断されます。遺伝子検査については、今のところ古典型EDSと血管型EDSが一定の条件の下で保険収載されています。
また、難病情報センターのページにおいて、2024年4月より13の病型分類についての診断基準が掲載されています。
どのような治療が行われるの?
今のところ、EDSを根本的に、つまり、遺伝子から治すような治療法は見つかっていません。そのため、必要な診療科で定期検診を受け、状況に応じてそれぞれの症状に合わせた対症療法が行われます。
例えば、関節の症状による痛みに対しては、鎮痛薬が使用されます。関節を保護するリハビリテーションが行われたり、補装具が使用されたりすることもあります。皮膚の裂傷が起きた場合は、慎重に縫合が行われます。皮膚や関節の症状の予防には、激しい運動を控えることやサポーターの装着などが有用です。
血管型EDSの場合、定期的に画像検査を受け、動脈病変が見つかった場合には、治療を受けます。治療はできる限り保存的に行われるよう慎重に検討されますが、進行性の場合には血管内治療や外科的治療が考慮されます。β遮断薬(セリプロロール)による動脈病変の予防も行われます。腸管破裂が起きた場合には、緊急手術が行われます。
EDSは、本人やご家族がこの病気についてよく理解し、医療関係者(主治医、リハビリ担当者など)、教育関係者(保育園や幼稚園、学校)と情報を共有することが大切です。緊急時、どこに、どのように受診するかなども、共有すべき大事な情報です。健康状態を記したカードを携帯しておくという方法もあります。また、靴もしっかりと安定したものを選ぶと、けがの予防につながります。
どこで検査や治療が受けられるの?
日本でエーラス・ダンロス症候群の診療を行っていることを公開している、主な施設は以下です。
- 札幌医科大学附属病院 遺伝子診療科
- 信州大学医学部附属病院 遺伝子医療研究センター
- 東京大学医学部附属病院 マルファン症候群センター
- 東京科学大学病院 生命倫理研究センター遺伝子診療科
- 慶應義塾大学医学部 臨床遺伝学センター
- 国立成育医療研究センター 遺伝診療科
- 公益財団法人榊󠄀原記念財団附属榊󠄀原記念病院 臨床遺伝科
- 長崎大学病院 ゲノム診療センター
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
エーラス・ダンロス症候群の患者会で、ホームページを公開しているところは、以下です。
参考サイト
- 難病情報センター
- 小児慢性特定疾病情報センター エーラス・ダンロス(Ehlers-Danlos)症候群
- MedlinePlus
- Genetic and Rare Diseases Information Center
- Malfait F. et al., The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017 Mar;175(1):8-26.
- Online Mendelian Inheritance in Man(R) (OMIM(R))
- 難病領域における検体検査の精度管理体制の整備に資する研究班(終了) 保険収載されている遺伝学的検査(D006-4)







