



どのような病気?
メープルシロップ尿症(MSUD)は、分岐鎖アミノ酸と呼ばれる3つのアミノ酸「バリン」「ロイシン」「イソロイシン」を体内で適切に処理できない遺伝性疾患で、先天代謝異常症と呼ばれる疾患の1つです。この病気を持つ乳児の尿が独特な甘いにおいであることが、病名の由来となっています。耳あかも同様のにおいがします。生まれてすぐの新生児期には、においが明らかでないこともあります。
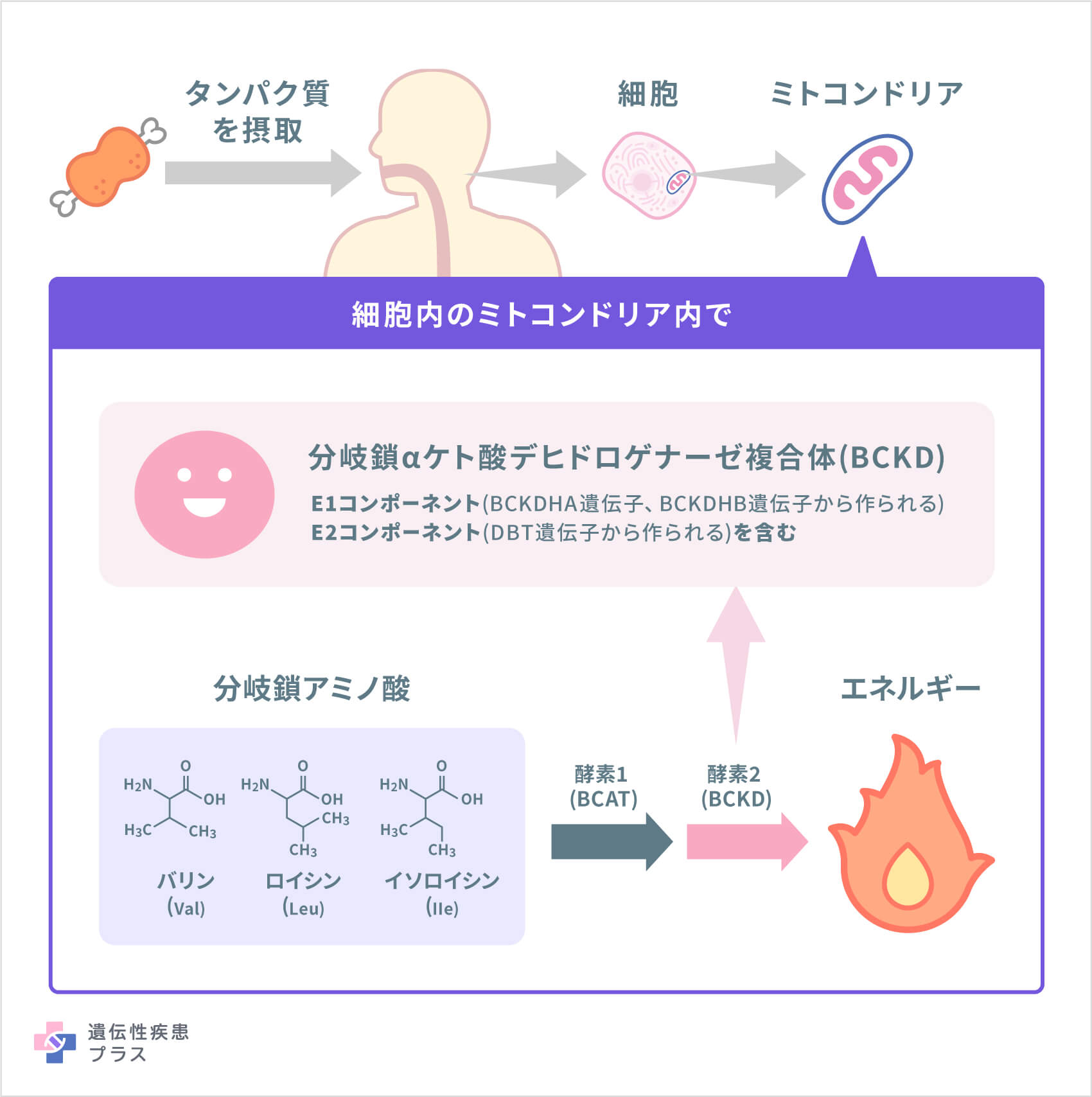
これら3つの分岐鎖アミノ酸は、タンパク質の構成要素で、多くの食品(特に牛乳、肉、卵などのタンパク質が豊富な食品)に含まれています。MSUDでは、「分岐鎖αケト酸デヒドロゲナーゼ複合体(BCKD)」という、酵素複合体(酵素の働きをもつタンパク質の集合体)が正常に働かなくなっているのですが、細胞のミトコンドリアという小器官で働いているこの酵素複合体は、食品から取り入れた分岐鎖アミノ酸を正しく分解・処理してエネルギーを取り出すために重要な役割を果たしています。そのため、MSUDでは体内にこれら3つのアミノ酸とその異常な代謝物質が増え、脳や臓器に損傷が起きます。
主な症状は、中枢神経障害とケトアシドーシス(ケトン体という物質が蓄積して体液が酸性になった状態)です。MSUDの中枢神経障害は、血液中のロイシンの濃度に相関することが知られています。血中ロイシン値が10~20 mg/dL(760-1,500μmol/L)では哺乳力の低下と嘔吐が見られます。ロイシン値が20 mg/dL(1,500μmol/L)以上では意識障害、筋緊張低下、けいれん、呼吸困難、後弓反張などが見られます。分岐鎖アミノ酸およびその中間代謝物である分岐鎖ケト酸(ロイシン由来の分岐鎖ケト酸はα ケトイソカプロン酸)の血中濃度が上昇すると、脳神経細胞の発達抑制が起こり、また、神経細胞に重要なミエリンが正常に合成できなくなります。さらに、高濃度のロイシンは、他のアミノ酸(中性アミノ酸)が脳に送られるのを抑制するため、それらのアミノ酸由来の神経伝達物質が欠乏すると考えられています。
MSUDには、生後1~2週間で発症する重篤な「古典型」、臨床症状がやや軽度な「中間型」、通常は無症状で急性増悪を起こす「間欠型」などの病型があります。最も一般的なのは古典型で、初期には元気がない、哺乳力低下、不機嫌、嘔吐などが見られます。進行すると、意識障害、けいれん、呼吸困難、筋緊張低下、後弓反張などが見られます。治療が遅れると重篤な神経後遺症が残ったり、呼吸不全で命を落としたりすることになります。間欠型や中間型は新生児期には無症状です。中間型は、幼児期以降に発症し、通常は軽度ですが、治療は必要です。間欠型は、感染症などをきっかけとして急に発症し、嘔吐や昏睡、発達の遅れなどが見られます。
| メープルシロップ尿症で見られる症状 |
|---|
99~80%で見られる症状 咽頭部の異常、声の異常、血漿中分岐鎖アミノ酸濃度上昇、全般的発達遅滞、知的障害、筋緊張低下、腱反射の低下、呼吸障害、発作 |
79~30%で見られる症状 運動失調症、片まひ |
割合は示されていないが見られる症状 脳浮腫、昏睡、乳児期の摂食困難、成長障害、幻覚、筋緊張亢進、低血糖、ケトーシス、乳酸アシドーシス、無気力、膵炎、嘔吐 |
MSUDは、新生児マススクリーニング検査の対象疾患として、厚生労働省が各自治体に勧奨している20の疾患のうちの1つです。早期発見・早期治療や、肝臓移植によって、症状が改善することが知られていますが、20の疾患の中で最も死亡率が高く、後遺症が残ることも少なからずあります。MSUDの日本における発生頻度は約50万出生に1人とされており、日本にMSUDの患者さんは100人程度いると推定されています。世界的に見ると、乳児の18万5,000に1人がこの病気であると推定されています。また、米国のある集団(Old Order Mennonite)では頻度が高く、新生児380人に1人の割合で発生すると推定されています。
MSUDは、国の指定難病対象疾患(指定難病244)、および、小児慢性特定疾病の対象疾患です。
何の遺伝子が原因となるの?
MSUDの原因遺伝子として、「BCKDHA」「BCKDHB」「DBT」の3つの遺伝子が見つかっています。BCKDHA遺伝子は19番染色体の19q13.2、BCKDHB遺伝子は6番染色体の6q14.1、DBT遺伝子は1番染色体の1p21.2という位置にそれぞれ存在します。
これら3つの遺伝子は、「分岐鎖αケト酸デヒドロゲナーゼ複合体(BCKD)」を構成するタンパク質の設計図となる遺伝子です。BCKDHA遺伝子からはBCKDの一部となる「E1αサブユニット」が、BCKDHB遺伝子からは「E1βサブユニット」が作られます。αサブユニット2つとβサブユニット2つが会合して、BCKDのE1コンポーネントと呼ばれる重要な部分を形成します。DBT遺伝子からは、やはりBCKDの重要な部分であるE2コンポーネントを構成するタンパク質が作られます。
これら3つの遺伝子のいずれかに変異があると、BCKD機能が低下また失われ、バリン、ロイシン、イソロイシンが正常に処理されなくなります。その結果、これらのアミノ酸とその異常な代謝物が体内に蓄積します。これらの物質が体内で高濃度になると、脳やその他の臓器に有害となり、メープルシロップ尿症の発症につながります。
BCKDにはE3コンポーネントもあり、これを構成するタンパク質の設計図となるDLD遺伝子に異常がある場合もMSUDと同様の症状が現れますが、E3は他の種類の酵素複合体(ピルビン酸脱水素酵素複合体、αケトグルタル酸脱水素酵素複合体)とも共通したコンポーネントです。そのため、DLD遺伝子は、高乳酸血症、αケトグルタル酸の上昇など、別の症状が見られる疾患(ジヒドロリポアミドデヒドロゲナーゼ欠損症、リー脳症)の原因遺伝子とされています。
これらの遺伝子変異によるMSUDは、いずれも常染色体劣性(潜性)遺伝形式で親から子へ遺伝します。人間が2本1セットで持っているそれぞれの原因遺伝子のうち、両親がともに1本ずつ変異を有していた場合、子どもは4分の1の確率で2本とも変異を有してMSUDになります。また、2分の1の確率で1本変異を有し発症はしない「保因者」となり、4分の1の確率でMSUDを発症せず保因者でもなく(変異した遺伝子を持たず)生まれます。

どのように診断されるの?
MSUDは、新生児マススクリーニング検査で発見されることがほとんどです。一方で、新生児マススクリーニング検査では間欠型を発見できないことがあるため、新生児期以降の発症に対して診断が行われる場合もあります。日本先天代謝異常学会による、MSUDの診断基準は以下のようになっています。
まず、血中・尿中アミノ酸分析において、バリン、ロイシン、イソロイシンの増加、アラニンの低下を認め、特に、血中ロイシン値が4mg/dL(300µmol/dL)以上であり、次の2つのどちらかが認められた場合、MSUDと診断されます。
- 尿中有機酸分析において、分岐鎖αケト酸、分岐鎖αヒドロキシ酸の増加を認めた場合。
- リンパ球・皮膚線維芽細胞・羊水細胞・絨毛細胞などを用いたBCKDの酵素活測定において活性低下(正常対照の20%以下)を認めた場合。※5%未満の場合は古典型、5~20%の場合は中間型あるいは間欠型と病型分類されます。
その他の診断の根拠となる検査として遺伝子解析も挙げられますが、BCKDを形成するそれぞれの酵素について解析が必要になるなどの理由から、診断には用いられていません。
どのような治療が行われるの?
新生児発症の急性期には、適切なカロリー(80kcal/kg以上)と電解質輸液、ビタミンB1(チアミン)投与(チアミン投与で症状が改善する病型もある)、タンパク質制限で治療が行われます。症状が改善しない場合には、肝移植や血液ろ過透析が行われる場合もあります。慢性期にはアミノ酸の制限食や特殊ミルクを使用していきます。
MSUDは進行性のものがあり、神経学的予後も良好とは言えませんが、早期診断・早期治療により、新生児期の初回急性増悪を抑えることができれば、良好な経過が期待されます。さらに肝臓移植により良好の経過が得られることが判明しています。
日常生活で、熱が出たり嘔吐・下痢を起こしたりしたときには、早めに医療機関を受診し、治療を受けましょう。
どこで検査や治療が受けられるの?
日本でメープルシロップ尿症の診療を行っていることを公開している、主な施設は以下です。
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
メープルシロップ尿症の患者会で、ホームページを公開しているところは、以下です。
参考サイト
- 難病情報センター
- 小児慢性特定疾病情報センター メープルシロップ尿症
- MedlinePlus
- Genetic and Rare Diseases Information Center
- Online Mendelian Inheritance in Man(R) (OMIM(R))







