



どのような病気?
メンケス病は、体の中の銅の吸収や利用がうまくいかないことにより、神経症状のほか、関節・臓器・血管などを支える結合組織などにさまざまな症状が引き起こされる遺伝性疾患です。ほとんどの場合男性で発症しますが、まれに女性でも報告があります。
メンケス病は、通常は生まれて数か月経ってから、まばらで縮れた頭髪、成長の遅れ、摂食障害、てんかん発作などの特徴や症状によって気が付かれます。毛髪の特徴は、短い、まばら、粗い、ねじれている、色素が薄いなどで、眉毛にも見られる場合があります。また、頬のたるみがあり、下膨れしたように見えることがあります。それ以外の症状としては、筋緊張低下、発達の退行や知的障害のほか、血管壁の異常、膀胱憩室、漏斗胸、臍ヘルニアや鼠径ヘルニア、めまいや失神などの自律神経失調症状、骨粗しょう症なども見られる場合があります。
多くの場合、症状は乳児期までに現れますが、小児期以降に発症する場合もあります。この病気は、肺炎、尿路感染などの感染症、血管破裂など重篤な症状が起こりやすく、幼児期までに命を落とす例も多いとされます。
また、この病気とよく似た病気としてオクシピタル・ホーン症候群があります。オクシピタル・ホーン症候群は、メンケス病の軽症型で小児期後期から青年期に診断されます。
メンケス病で見られる症状 |
|---|
高頻度に見られる症状 縮毛、髪の色素が少ない、毛髪が少ない、小頭症、口蓋(口の中の上部分)の形態異常、漏斗胸、腹壁の筋肉(おなかを取り囲む筋肉)の形成不全、乾燥肌、皮膚過伸展(異常に伸縮する)、筋緊張亢進、筋緊張低下、ヘルニア、鼠径(そけい)ヘルニア、頭蓋内出血、関節の可動性亢進(動きすぎる)、乳児期の摂食困難、てんかん発作、けいれん、倦怠感、発達の退行 |
良く見られる症状 突き出した後頭部、目立つ頬部、小顎症、仮面様の顔立ち(表情が乏しい)、胸(胸囲)が狭い、厚い皮膚、皮膚の非定型瘢痕(はんこん、傷跡)、筋力低下、ウォルム骨(頭蓋縫合線内にある余分な骨)が見られる、骨幹端の異常、外骨腫(骨の外側に突き出た良性腫瘍)、静脈機能不全、頸動脈の形態異常、動脈狭窄、新生児黄疸、吸収不良(栄養素を吸収する能力の障害)、吐き気と嘔吐、行動異常、知的障害 |
しばしば見られる症状 足根骨癒合、長骨の湾曲、軟骨石灰化、骨髄炎、骨粗しょう症、骨折を繰り返す、血腫の自然発生、消化管出血、膀胱憩室、敗血症、舞踏病、子宮内胎児発育遅延 |
メンケス病は、難病情報センターの情報によれば、平成22~23年度の国内調査において、過去10年間に62人の患者さんが見つかっており、そこから推定される発症率は、男児出生約12万人あたり1人となっています。
メンケス病は、国の指定難病対象疾病(指定難病169)、およびメンケス(Menkes)病として小児慢性特定疾病の対象疾患となっています。
何の遺伝子が原因となるの?
メンケス病は、X染色体のXq21.1領域に存在するATP7A遺伝子の変異が原因で引き起こされます。ATP7A遺伝子は、体内の銅を調節するために重要なタンパク質「銅輸送ATPase」の設計図となります。別の遺伝性疾患である、ウィルソン病の原因遺伝子ATP7B遺伝子が設計図となる銅輸送ATPase2と似た機能をもっていますが、銅輸送ATPase2が主に肝臓で機能するのに対し、ATP7A遺伝子が設計図となる銅輸送ATPaseは、主に肝臓以外で働きます。
銅は、多くの細胞の機能にとって必須のミネラルの一つですが、必要な量はわずかで過剰に存在すると有毒となります。銅輸送ATPaseは食べ物を消化し吸収する過程の中で、小腸から吸収された銅を各細胞内で制御するために働きます。この銅輸送ATPaseは、細胞の中のゴルジ体と呼ばれる細胞小器官に存在しており、細胞に送られてきた銅を受け取ると骨・皮膚・毛髪・血管・神経系などの構造や機能に重要な酵素に対し、その酵素の働きに必要な銅を付加します。また銅輸送ATPaseは、細胞の中に銅が増えすぎた時には細胞膜に移動し細胞の外に銅を出す働きをします。
ATP7A遺伝子の変異によって、銅輸送ATPaseの働きが失われると、体の中で銅がうまく分配されなくなります。その結果、小腸や腎臓などでは銅が蓄積し、脳やその他の組織では銅が不足します。また、骨や皮膚、毛髪、血管、神経などの構造や機能に必要な酵素に銅が付加されず、これらの酵素が機能できなくなることで、メンケス病の症状が引き起こされると考えられています。

メンケス病は、X連鎖劣性(潜性)遺伝形式で遺伝します。この遺伝形式では、X染色体を一つしか持たない男性(XY)の場合、変異のある遺伝子のコピーを一つ受け継ぐことで発症します。また、男性(父親)がこの変異を持つ場合には、息子には変異が受け継がれませんが、娘にはその変異のあるX染色体が受け継がれます。女性(XX)の場合は、変異のあるX染色体を持っていても、多くの場合は2本の染色体のうちもう一方の正常な遺伝子が機能を補うため、発症せず保因者となります。女性がこの変異を持つ場合には50%の確率でその原因遺伝子が存在するX染色体が娘または息子に受け継がれます。両親はこの病気の変異を持っておらず、新しく発生した変異が原因でこの病気を発症する(孤発例)場合もあります。
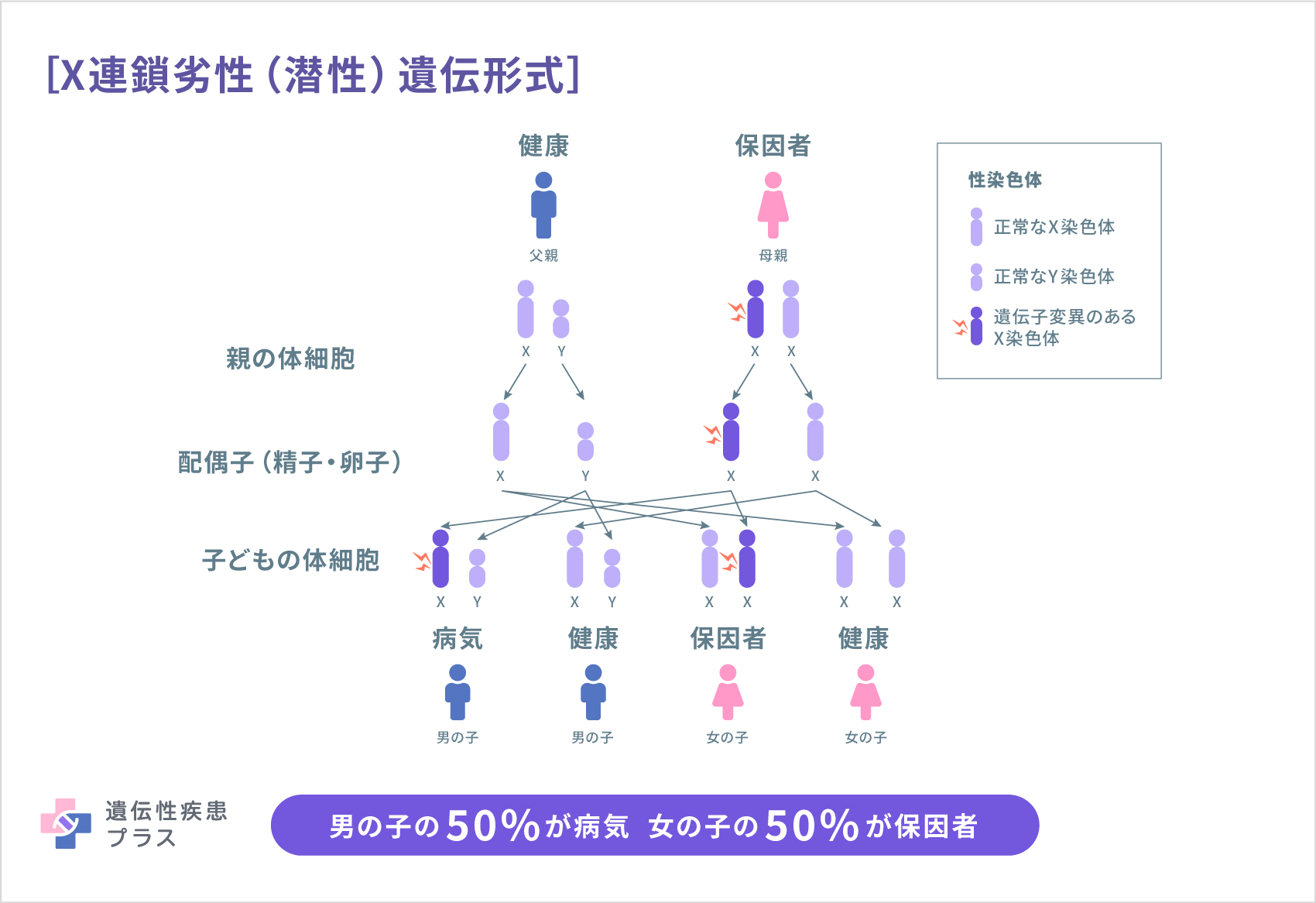
どのように診断されるの?
メンケス病の診断基準は、主な症状として頭髪異常(少ない毛、縮れ毛、色素減弱)が見られ、他に、
- 重度の中枢神経障害(著明な発達遅延)
- 難治性けいれん
- 硬膜下出血
- 骨粗しょう症、骨折
- 筋力低下
のうち1つ以上が見られること、加えて、血液検査または画像検査において、
1)血清銅値:30µg/dL以下でセルロプラスミン値:15mg/dL以下
2)血清乳酸・ピルビン酸の上昇
3)MRAで血管蛇行、MRIで脳萎縮・硬膜下出血のいずれか
4)骨粗鬆症、骨折のいずれか
5)膀胱憩室
の、1)~5)のうち1)を含む2つ以上が確認されること、
さらに、ミトコンドリア遺伝子異常症ではないと診断され(鑑別診断)、特殊検査によって培養皮膚線維芽細胞の銅濃度の高値が見られるか遺伝学的検査によりATP7A遺伝子に変異が認められることでこの病気であると診断されます。
特殊検査による培養皮膚線維芽細胞の銅濃度、遺伝学的検査のATP7A遺伝子変異が認められない場合においても、血液検査または画像検査において、1)と2)の両方、または1)と3)~5)のうち2つ以上が認められた場合には、この病気の可能性があると診断されます。
どのような治療が行われるの?
メンケス病において、根本的にこの病気を治療する方法はまだ確立されていないため、症状に応じた対症療法が行われます。保険適用はありませんが、新生児期に、神経症状が現れる前にヒスチジン銅を皮下注射する治療を行うことで神経障害に対する予防や軽減効果が期待できるとされます。この治療は、神経症状が現れた後では改善が見られません。また、この治療法では、結合組織の異常は改善されないため、血管異常、感染症、膀胱憩室などには引き続き注意が必要になります。
どこで検査や治療が受けられるの?
日本でメンケス病の診療を行っていることを公開している、主な施設は以下です。
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
難病の患者さん・ご家族、支えるさまざまな立場の方々とのネットワークづくりを行っている団体は、以下です。
参考サイト
- 難病情報センター
- 小児慢性特定疾病情報センター メンケス(Menkes)病
- MedlinePlus
- Genetic and Rare Diseases Information Center
- Online Mendelian Inheritance in Man(R) (OMIM(R))






