



どのような病気?
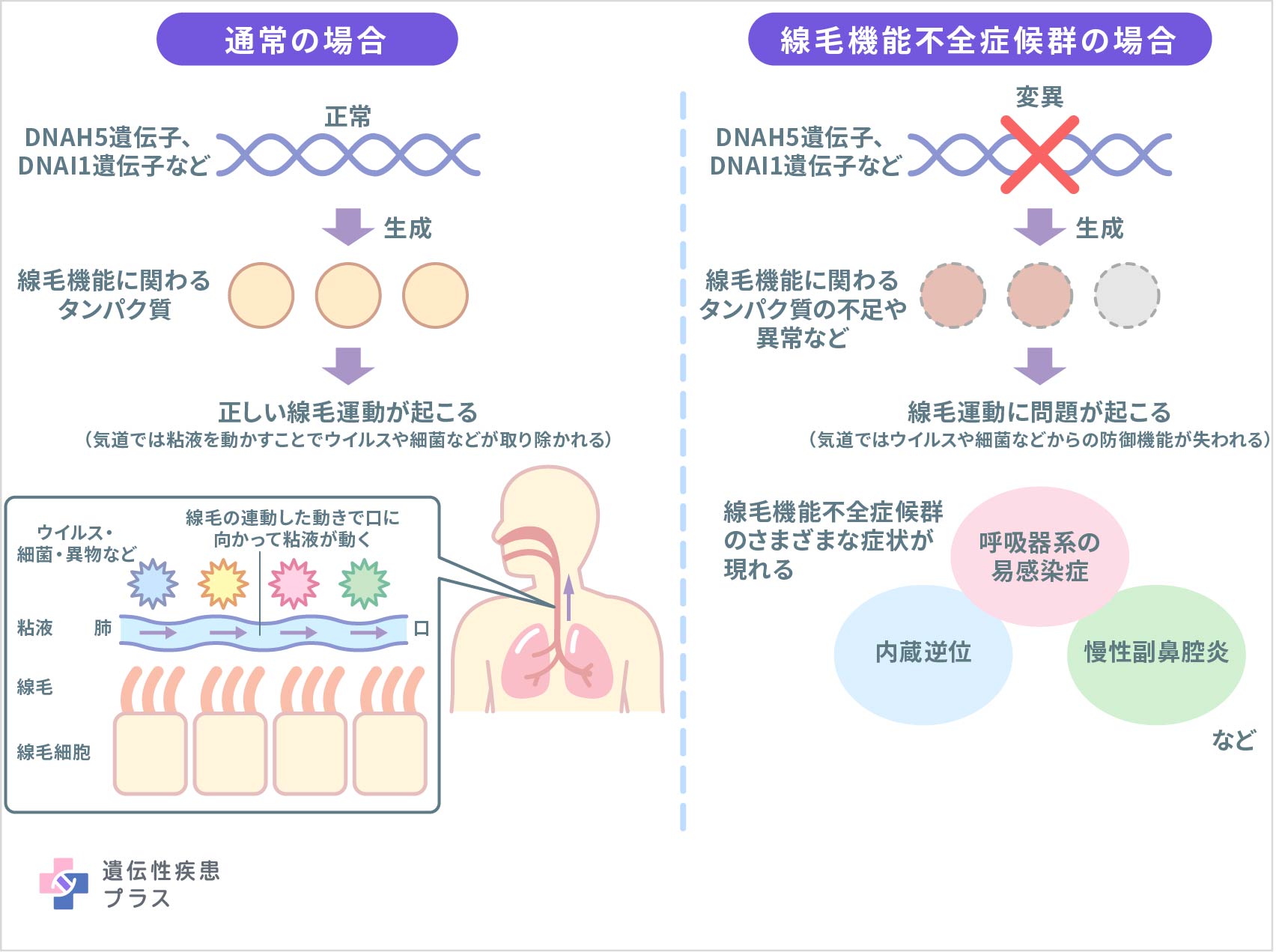
線毛機能不全症候群は、「線毛」の機能が失われることで、さまざまな症状が引き起こされる遺伝性疾患です。
線毛は、気道の中で粘液を動かし、侵入してきた異物を除去する役目を持っています。気道とは、肺につながる細長い管で、呼吸を行う際に空気の通り道となります。気道で空気に触れる細胞の表面には多くの線毛が存在し、線毛は連動して口の方に向かって動くことにより粘液を動かしています。そうしてできた粘液の流れには、小さい埃のような異物、細菌、痰などを除く役割があり、それによって気道の内部は清浄化されます。
線毛機能不全症候群では、この線毛の運動が遺伝子の変異によって損なわれることによって、耳(中耳、耳管)、鼻(副鼻腔)、のど(咽頭)などを含む呼吸器系で防御機能が低下し、病原菌などに感染しやすくなります。線毛は気道以外の臓器にも広く存在しており、耳の感染症は、内耳内の線毛に関連している可能性もあります。
この病気を持って生まれた赤ちゃんは、多くの場合呼吸に問題を抱えており、数日から2~3週の酸素投与を必要とすることもあります(呼吸窮迫・こきゅうきゅうはく)。乳幼児期以降は、慢性的な鼻水(鼻漏)や痰のある咳(湿性咳)が見られ、運動や深呼吸などで簡単に咳が出ることもあります。他にも、粘り気のある痰、肺炎、長引き繰り返す気管支炎、のほか、副鼻腔炎、中耳炎、慢性的な鼻づまり(鼻閉)、膿性鼻汁、耳鳴り、難聴なども引き起こされることがあります。
慢性的な呼吸器感染症により気管支が傷つくと、気管支が拡張して戻らない気管支拡張症と呼ばれる状態となり、悪化することで喘鳴(ぜんめい、ゼイゼイ、ヒューヒューのような音がする呼吸)、呼吸困難、ばち状指(指先が膨らんで太鼓のばちのように変形する)、などが見られる場合もあります。
線毛は生殖器系にもみられ、男性では精子の鞭毛運動、女性では卵管の線毛運動に問題が起こることにより不妊症につながる場合もあります。その他にも、脳内の異常な線毛運動が原因となって脳内に体液が蓄積する水頭症や、嗅覚障害(嗅細胞の線毛異常)、視覚障害(網膜色素変性症)などの合併症も見られます。
また、胚の発生時において、体の左側と右側が決定されるのにも線毛の動きは重要です。そのため、この病気ではおなかの中にいる胎児期において、左右が適正に判別できない症状が認められ、約半数で完全内臓逆位(内臓の全てが左右逆に位置している)となる「カルタゲナー(Kartagener)症候群」とよばれる病型をとることがあります。また、それ以外の約1割程度の患者さんにおいても、心臓、肝臓、腸、脾臓などの構造や、位置が通常とは異なるといった症状が見られることがあります。
線毛機能不全症候群は、以前、線毛不動症候群とも呼ばれていましたが、線毛の動きが全くない場合だけでなく、動きが弱い場合や協調した動きが損なわれ不揃いとなっている場合なども含め、現在ではこの名前が使われています。
線毛機能不全症候群で見られる症状 |
|---|
良く見られる症状 慢性鼻炎、慢性副鼻腔炎、再発性副鼻腔肺感染症、鼻詰まり、鼻ポリポーシス、再発性中耳炎、慢性中耳炎、湿性咳、喀痰の異常、呼吸器感染、精子の運動性異常、男性不妊症、新生児期の呼吸困難 |
しばしば見られる症状 ばち指、気道閉塞、無気肺(肺の一部または全部がつぶれて空気が入っていない状態)、気管支拡張症、気管支血管周囲間質の肥厚、肺の位置の異常、完全内臓逆位、異常な心臓の形態、心血管系の形態異常、泌尿生殖器の異常、骨格の異常、伝音難聴、聴覚障害、発話と言語発達の遅延、子宮外妊娠、女性不妊症、中枢神経系の形態異常、再発性のマイコバクテリア感染症、喘鳴 |
まれに見られる症状 水頭症、脳室拡大、桿体錐体ジストロフィー、肺静脈還流異常、左上大静脈遺残、両大血管右室起始症、大動脈の転位、心房の位置の異常、心房不定位、多脾症、無脾症、腸回転異常症、呼吸不全 |
小児慢性特定疾病情報センターによれば、線毛機能不全症候群の発症頻度は出生1万人~4万人に1人であると推定されています。この病気は未診断や未登録の患者さんがまだ多くいるとみられており、正確な患者数はわかっていません。
線毛機能不全症候群は、「線毛機能不全症候群(カルタゲナー症候群を含む。)」として国の指定難病対象疾病(指定難病340)、および「線毛機能不全症候群(カルタゲナー(Kartagener)症候群を含む。)」として小児慢性特定疾病の対象疾患となっています。
何の遺伝子が原因となるの?
線毛機能不全症候群における線毛運動の異常には、さまざまな遺伝子の変異が関わっている可能性があります。
線毛が正常な構造と機能を発揮するためには、多くの異なる遺伝子が関与していることがわかっており、これらの遺伝子の中には、ダイニンと呼ばれる線毛の内部構造を形成して線毛が曲がるために必要なタンパク質の設計図となる遺伝子も含まれ、これらの遺伝子の変異によって線毛の連動した動きが失われます。線毛の動きは体内において多くの臓器や組織が正常に機能するために必要であることから多様な症状を引き起こします。
これまで明らかになっている原因遺伝子には、ダイニンに関わる変異が多く知られており、DNAH5、DNAI1、DNAH11、TXNDC3、DNAI2などが同定されています。中でもDNAI1およびDNAH5遺伝子の変異は、この病気の患者さんの3割程度を占めているとされます。
他にもKTU、RPGR、OFD1、RSPH9、RSPH4A、RPGR、CCDC103、CCDC39、CCDC40、CCDC65、CFAP298、DNAAF1、DNAAF11、DNAAF2、DNAAF3、DNAAF4、DNAAF5、DNAH8、DNAL1、DRC1、HYDIN、NME8、ODAD1、ODAD2、RSPH1、SPAG1、ZMYND10など多数の遺伝子変異が原因として報告されており、欧州の希少疾患情報サイト(orphanet)によると、この病気の原因として、これまでに46の異なる遺伝子の変異が知られています。一方で、多くの患者さんにおいて原因となる遺伝子が不明であることや、線毛を構成するタンパク質はおよそ250種類であると言われることから、今後も新しい遺伝子変異が発見される可能性があります。
この病気は主に、常染色体劣性(潜性)遺伝形式で遺伝します。この遺伝形式では、それぞれの細胞に2つずつ存在する遺伝子の両方に変異が生じることにより発症します。両親は変異のある遺伝子と変異のない遺伝子を1つずつ持つ保因者で、通常はこの病気を発症していません。

どのように診断されるの?
線毛機能不全症候群では、主要な症状として、1)難治性の慢性湿性咳嗽(痰のある咳)、2)難治性の副鼻腔炎、3)完全内蔵逆位があります。他にも、肺聴診でcrackles&rhonchiと呼ばれる肺雑音を認める、肺機能検査において閉塞性換気障害を認める、胸部単純X線で無気肺や気管支壁肥厚像を認める、などがこの病気が疑われる重要な所見となります。
スクリーニング検査としては、鼻の中(下鼻甲介)にサッカリンの粒を置いて咽頭で甘味を感じるまでの時間を計測するサッカリンテストや、鼻腔内の一酸化窒素(NO)濃度を測定し異常低値となるかを調べるなどの検査が行われることがあります。
確定診断は、先天的な線毛運動の異常について確認が行われます。通常は、鼻粘膜や気管支から線毛上皮細胞を取り線毛運動があるかどうかを顕微鏡で確認する方法がとられます。運動に異常が見られた場合にはさらに線毛の微細構造を詳しく調べ、異常が認められればこの病気と診断されます。
DNAH5やDNAI1など高頻度の遺伝子異常について、遺伝学的診断が行われることもあります。
どのような治療が行われるの?
この病気の根本的な治療はまだなく、症状に合わせた対症療法がとられます。
この病気の長期的な管理の中心として胸部理学療法があり、病変部を上にした体位で振動を与え、咳を起こさせるという方法を毎日続けます。薬物療法としては痰を除く(去痰薬)や、気管支拡張薬などがあります。抗菌薬が選択されることもあります。
感染症の悪化に対して早期に対応し、気管支拡張症や呼吸不全への進展を遅らせることが重要であるため、理学療法や薬物両方の継続が予後に影響するとされます。再発性の中耳炎は、適切な治療が行われないと難聴につながることもあります。
こうした治療は、耳鼻咽喉科、眼科など関連する診療科が連携して行われます。
どこで検査や治療が受けられるの?
日本で線毛機能不全症候群の診療を行っていることを公開している、主な施設は以下です。
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
難病の患者さん・ご家族、支えるさまざまな立場の方々とのネットワークづくりを行っている団体は、以下です。
米国の線毛機能不全症候群の患者支援団体(財団)で、ホームページを公開しているところは、以下です。
参考サイト
- 小児慢性特定疾病情報センター 線毛機能不全症候群(カルタゲナー(Kartagener)症候群を含む。)
- MedlinePlus
- Genetic and Rare Diseases Information Center
- orphanet






