



どのような病気?
レット症候群は、乳幼児期に症状が現れる発達障害で、患者さんはほとんどが女性です。その理由は、原因遺伝子に関連していると考えられています。症状や病気の程度は、人によって、また、年齢によってさまざまですが、約8割以上の患者さん(典型例)は、生後半年くらいまで目立った症状なく過ごします。しかし、日中の睡眠時間が長く、外界からの刺激に対する反応が薄いため、「おとなしく、よく眠る、手のかからない子」という表現が当てはまります。生後半年を過ぎると、発達が止まって言語と運動能力が急速に低下します。症状の経過は、大きく次の4つのステージに分けて考えることができます。
第1期:発達停滞期(生後6~18か月、数か月続く)
生後6か月過ぎくらいから、体が柔らかい、四つんばいや歩行などの運動の遅れ、外界への反応が乏しい、視線が合いにくいなどの自閉症状が出てきます。また、喃語(なんご)や意味のある言葉が出ていても、その後言葉の発達がみられなくなります。
第2期:退行期(1~4歳から数か月続く)
これまで出来ていた運動、言語が急激に出来なくなります。おもちゃやスプーンを持てなくなるなど、今まで使っていた手の運動が上手に出来なくなり、特有な手の動き(手もみ、手絞り、手を口に持っていく、片手で胸を叩くなどを繰り返す)が出現します。これまで出来ていた四つんばいや歩行も難しくなります。周囲とのコミュニケーションができなくなり、それまでしゃべっていた言葉が出なくなったりもしてきます。
第3期:仮性安定期(2歳~10歳に始まり、数年から数十年続く)
急激な退行の後に、症状が安定します。視線もよく合うようになります。特有な手の動きや呼吸の異常、歯ぎしりなどがよくみられます。約40~80%でてんかんが認められ、筋緊張が次第に亢進してきます。
第4期:晩期機能低下期(10歳~)
手足を使って動くことがほとんどないために細くなり、車いすを使うようになります。筋緊張がさらに亢進し、ジストニアと呼ばれる不随意運動が著明になります。脊柱側弯、運動機能の低下がみられ、骨折のリスクも増えます。
| レット症候群でみられる症状 |
99~80%でみられる症状 呼吸パターンの異常、言語発達がみられない、発達の退行、全般的発達遅延、甲高い泣き声、進行性小頭症、常同的手絞り運動 |
79%~30%でみられる症状 動作緩慢、歩行困難、ジストニア、脳波異常、成長障害、肢体不自由、けいれん発作、骨格筋萎縮 |
29%~5%でみられる症状 自律神経系の生理異常、アジテーション(興奮、不穏)、胆のう炎、高アンモニア血症、歩行不能、脳脊髄液乳酸の増加、血清乳酸の増加、血清レプチンの増加、血清ピルビン酸の増加、乳児の筋緊張低下、脊柱側弯症、睡眠障害 |
4%~1%でみられる症状 先天性小頭症 |
割合は示されていないがみられる症状 心電図T波の異常、歯列の異常、無呼吸、自閉症様行動、歯ぎしり、悪液質、大脳皮質の萎縮、便秘、認知症、歩行失行、歩行運動失調、胃食道逆流症、知的障害(IQ20未満)、断続的な過呼吸、脊柱後湾症、運動機能の後退、出生後の小頭症、心電図間隔の延長、短い足、低身長、けいれん性まひ、体幹運動失調 |
日本にレット症候群の患者さんは、1,000人程度いると推定されています。発症頻度は10万人の女児に8~9人くらいと報告されています。また、世界のどの国でも発症すると考えられており、大きな人種差は無い可能性があります。
レット症候群は、国の指定難病対象疾患(指定難病156)、および、小児慢性特定疾病の対象疾患です。
何の遺伝子が原因となるの?
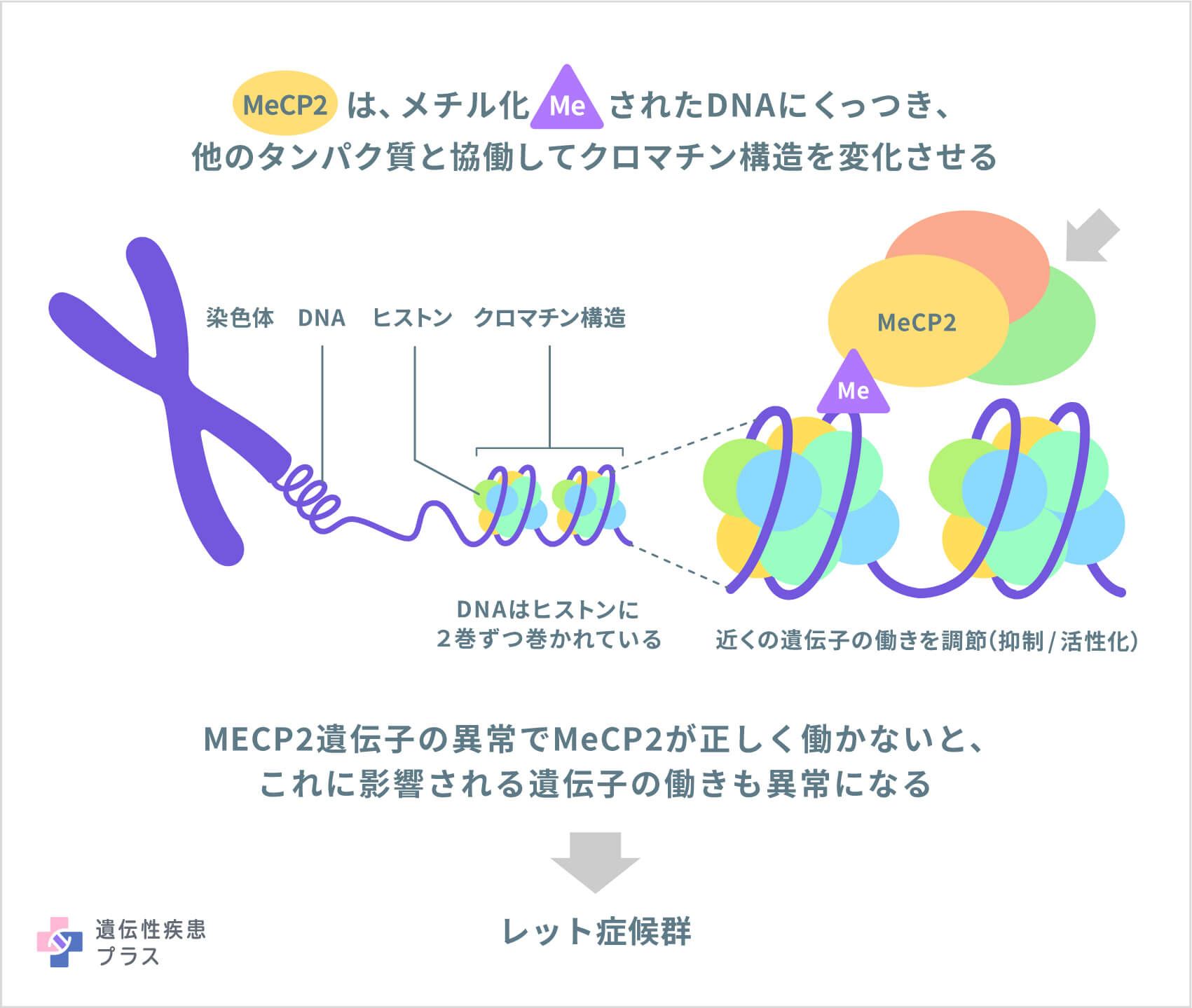
レット症候群の原因として、X染色体のXq28という位置に存在する「MECP2遺伝子」が、これまでに見つかっています。典型例の患者さんの約90%で、この遺伝子に変異があることがわかっています。MECP2遺伝子は、メチルCpG結合タンパク質2(Methyl-CpG-binding protein 2、MeCP2)というタンパク質の設計図となる遺伝子です。MeCP2は、「メチル化」と呼ばれる化学修飾を受けているDNAに結合し、他のタンパク質たちと協働して、「クロマチン」と呼ばれる染色体の構造を変化させることにより、その近くにある遺伝子の働きを調節する役割を担っています。
MeCP2タンパク質は、体全体の細胞に存在しますが特に脳細胞に豊富に存在し、MeCP2によって調節されることが知られている遺伝子の多くは、神経系の発達と正常な脳機能、特にシナプスの維持に重要な役割を果たしています。また、MeCP2は、遺伝子からタンパク質が作られる過程で「メッセンジャーRNA(mRNA)」という分子に、「選択的スプライシング」と呼ばれる大切なプロセスが起こるためにも重要であることがわかっています。脳では、選択的スプライシングはニューロン同士が正常にコミュニケーションするために重要で、脳細胞の機能にも必要な場合があります。
レット症候群の原因となるMECP2遺伝子の変異は、正常なMeCP2が作られなくなったり、作られるMeCP2の量が少なすぎたりするような変異で、これにより、脳細胞が正しく機能できなくなると考えられています。レット症候群の重症度は、MECP2遺伝子のどこにどのような変異が入っているか、などが関わっていると考えられています。
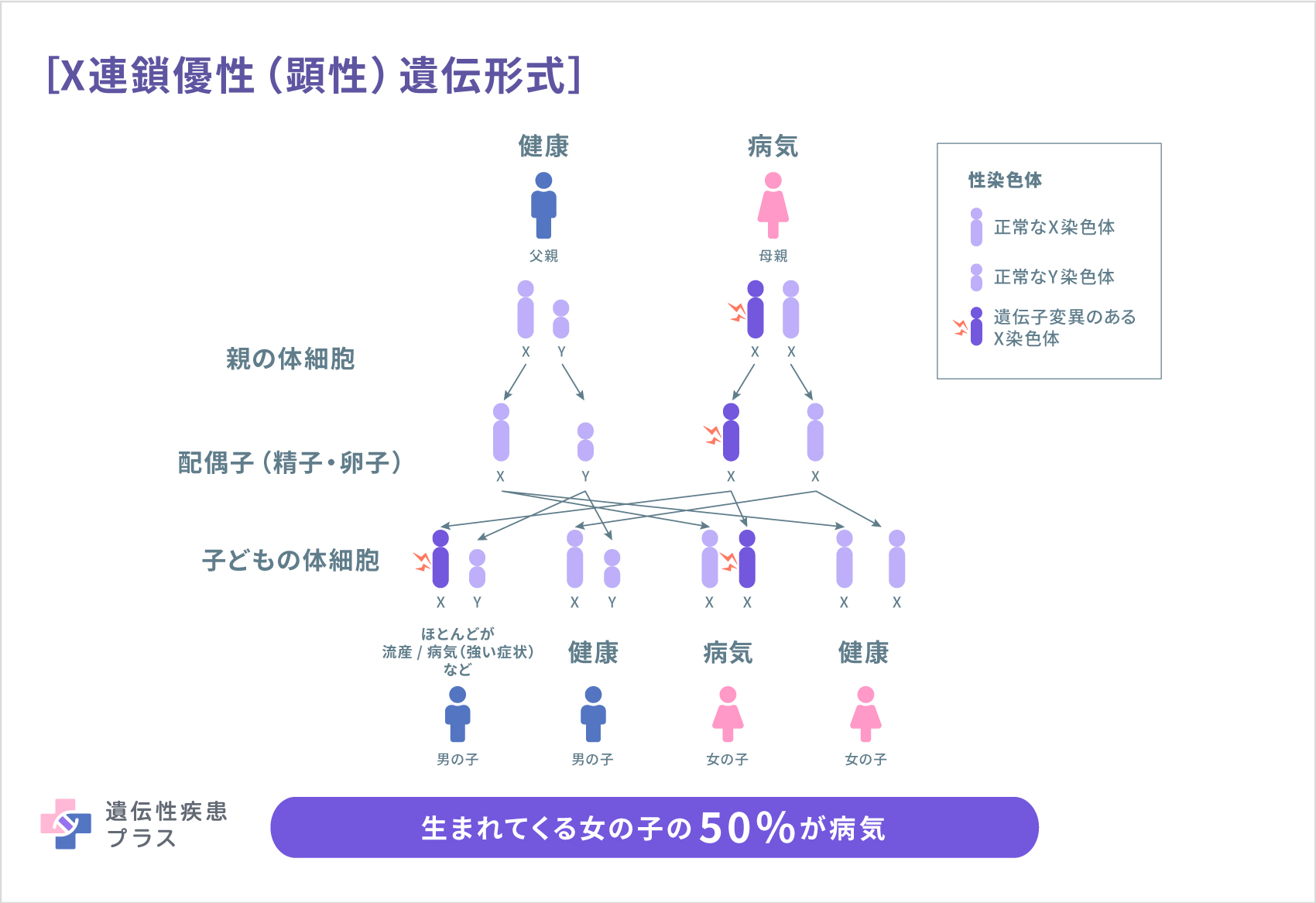
MECP2遺伝子の変異によるレット症候群は、X連鎖優性(顕性)遺伝と呼ばれる形式で遺伝します。人間は、性染色体と呼ばれる染色体を2本持っています。性染色体にはX染色体とY染色体の2種類があり、XYの組み合わせは男性、XXの組み合わせは女性になります。MECP2遺伝子は、X染色体に存在する遺伝子です。
男性が1本のみ有するX染色体のMECP2遺伝子に変異があると、正常なMeCP2タンパク質は全く作られないため、ほとんどが生まれることなく流産となります。
一方女性の場合、X染色体を2本持っているので、1本が異常でも、もう1本のMECP2遺伝子が正常に働き、機能を補完します。そのため、死んでしまうことはなく、レット症候群として生まれてきます。したがって、レット症候群は、一般に母親から娘に遺伝する形か、新生変異(その子どもで新たに起きた変異)となりますが、レット症候群の遺伝子変異は、大部分が両親には変異がなく新生変異で、「家系」がみられる例はほとんどありません。レット症候群の患者さんが子どもを持った場合には、母親から娘にこの病気が遺伝する確率は、50%です。つまり、レット症候群の母親から生まれる子どもは、レット症候群の女児:レット症候群でない女児:レット症候群でない男児=1:1:1となります。
極めてまれですが、MECP2遺伝子に変異のあるX染色体が選択的に不活化されて母親保因者から遺伝したという報告や、精子や卵子の前駆細胞である生殖細胞が「モザイク」という状態(MECP2遺伝子変異をもつものともたないものが混在している)になっている親から遺伝したという報告もあります。
その他、非典型例として、CDKL5遺伝子の異常や、FOXG1遺伝子の異常などが見つかっていますが、これらは、同じような兆候や症状がみられるものの、原因遺伝子も違うことなどから、レット症候群とは別の病気と考えられるようになってきています。
どのように診断されるの?
レット症候群には、医師がレット症候群と診断するための「診断基準」があります。したがって、病院へ行き、必要な問診や検査を受けた後、主治医の先生がそれらの結果を診断基準に照らし合わせ、結果的にレット症候群かそうでないかの診断をすることになります。
典型例の場合には、乳幼児期~小児期早期に「目的のある手の運動機能を習得した後に、その機能を部分的、あるいは完全に喪失する」「音声言語を習得後に、その機能を部分的、あるいは完全に喪失する」「歩行異常(歩行障害、歩行失行)」「手の常同運動(手をねじる・絞る、手を叩く・鳴らす、口に入れる、手を洗ったりこすったりするような自動運動)」の4つ全ての症状がみられ、「明らかな原因のある脳障害(周産期・周生期・後天性の脳障害、神経代謝疾患、重度感染症など)による脳損傷」「生後6か月までに出現した精神運動発達の明らかな異常」がみられず、さらに、アンジェルマン症候群、ピット・ホプキンス症候群、自閉症スペクトラム症(障害)などの発達障害ではなく、回復期や安定期が後続する退行期がある場合に、レット症候群と確定診断されます。
非典型例の場合には、「目的のある手の運動機能を習得した後に、その機能を部分的、あるいは完全に喪失する」「音声言語を習得後に、その機能を部分的、あるいは完全に喪失する」「歩行異常(歩行障害、歩行失行)」「手の常同運動(手をねじる・絞る、手を叩く・鳴らす、口に入れる、手を洗ったりこすったりするような自動運動)」の4つのうち2つがみられ、「明らかな原因のある脳障害(周産期・周生期・後天性の脳障害、神経代謝疾患、重度感染症など)による脳損傷」「生後6か月までに出現した精神運動発達の明らかな異常」の両方がみられ、「覚醒時の呼吸異常」「覚醒時の歯ぎしり」「睡眠リズム障害」「筋緊張異常」「末梢血管運動反射異常」「側弯・前弯」「成長障害」「小さく冷たい手足」「不適切な笑い・叫び」「痛覚への反応の鈍麻」「目によるコミュニケーション、じっと見つめるしぐさ」のうち5つ以上がみられ、さらに、アンジェルマン症候群、ピット・ホプキンス症候群、自閉症スペクトラム症(障害)などの発達障害ではなく、回復期や安定期が後続する退行期がある場合に、レット症候群と確定診断されます。
MECP2遺伝子、CDKL5遺伝子、FOXG1遺伝子の遺伝子検査も行われます。
どのような治療が行われるの?
今のところ、レット症候群を根本的に、つまり、遺伝子から治すような治療法は見つかっていません。そのため、現時点ではその人の症状に合わせて対症療法がおこなわれます。
運動に対しては理学療法や作業療法、言語に対しては言語療法がおこなわれます。長時間続くけいれんが起きないように薬を調整します。便秘が多いので、薬を使用することもあります。食べ物を飲み込むのに時間がかかるので、食事はむせないように、トロミをつけるなどの工夫をします。誤嚥性肺炎にも気を付けます。骨折がやや多いことが報告されているので、定期的に骨密度などを測定し、活性型ビタミンDやカルシウムを摂取する場合もあります。また、不整脈が出ることが報告されているので、毎年1回くらい、心電図の定期検診を受けます。側弯が進行しないように、整形外科に相談し、コルセットを作成することもあります。歯ぎしりのため、歯がすり減ったりするので、歯科治療を受けたり、歯肉炎、虫歯などの予防をしたりします。その他、水泳や音楽療法などが試みられています。
インスリン様成長因子(IGF-1)の皮下注射、その他の新規の治療薬開発も、世界で始まっています。また、2019年10月には、認定NPO法人レット症候群支援機構は自治医科大学と共に「遺伝子治療プロジェクト」を立ち上げました。レット症候群の遺伝子治療の実現に向け、自治医科大学の遺伝子治療研究チームによる研究が進められています。
どこで検査や治療が受けられるの?
日本でレット症候群の診療を行っていることを公開している、主な施設は以下です。
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
レット症候群の患者会・支援団体で、ホームページを公開しているところは、以下です。
参考サイト
- 難病情報センター
- 小児慢性特定疾病情報センター レット(Rett)症候群
- 国立精神・神経医療研究センター レット症候群
- Genetic and Rare Diseases Information Center







