



どのような病気?
アッシャー症候群は、難聴に網膜色素変性症を伴う疾患です。難聴は先天性が多く、その程度は中等度~重度が多くを占めます。網膜色素変性は遅発性に発症し、徐々に視野狭窄が進行して社会的失明(矯正視力<0.1程度)となることがあります。アッシャー症候群は症状の程度と発症時期によって3つのタイプに分類されます。
タイプ1:生まれつき重度の難聴がみられます。前庭機能(前庭は内耳にあるバランス感覚などに関わる器官)の障害を伴う場合が多く、目の症状としては、暗いと視力が低下する夜盲は10歳前後から発症します。タイプ1はアッシャー症候群の約4割を占めます
タイプ2:生まれつき高音障害型難聴(高い音ほど聞こえにくい難聴)がみられます。夜盲は思春期以降よりみられることが多いとされています。また、前庭機能は正常である場合が多いです。タイプ2はアッシャー症候群の約6割を占めます。
タイプ3:進行性の難聴が特徴です。前庭機能障害の有無や夜盲の発症時期はさまざまです。アッシャー症候群のうちタイプ3は3%未満とされています。
アッシャー症候群では、目および耳を中心として、さまざまな症状が見られます。
アッシャー症候群で見られる症状 |
|---|
高頻度に見られる症状 網膜電位異常、網膜色素沈着異常、盲目、夜盲症、進行性視覚障害、感音性難聴(内耳や聴神経の異常による難聴)、感覚神経障害、前庭部機能不全、視野欠損、視覚障害 |
よく見られる症状 運動失調、白内障、認知障害、遠視、近視 |
しばしば見られる症状 心血管系機能異常、歯のエナメル形態異常、歯の色素異常、不安症、小脳形成不全、乱視、う歯(虫歯)、皮質性小脳萎縮、不妊、うつ、幻覚、聴覚過敏、肥大型心筋症、小歯症、筋障害、眼振(意思と関係なく眼球が動くこと)、精神症、耳鳴り |
アッシャー症候群の有病率は10万人あたり4~17人とされています。国内の調査でも10万人あたり6.7人と推測されています。
アッシャー症候群は指定難病対象疾病(指定難病303)です。
何の遺伝子が原因となるの?
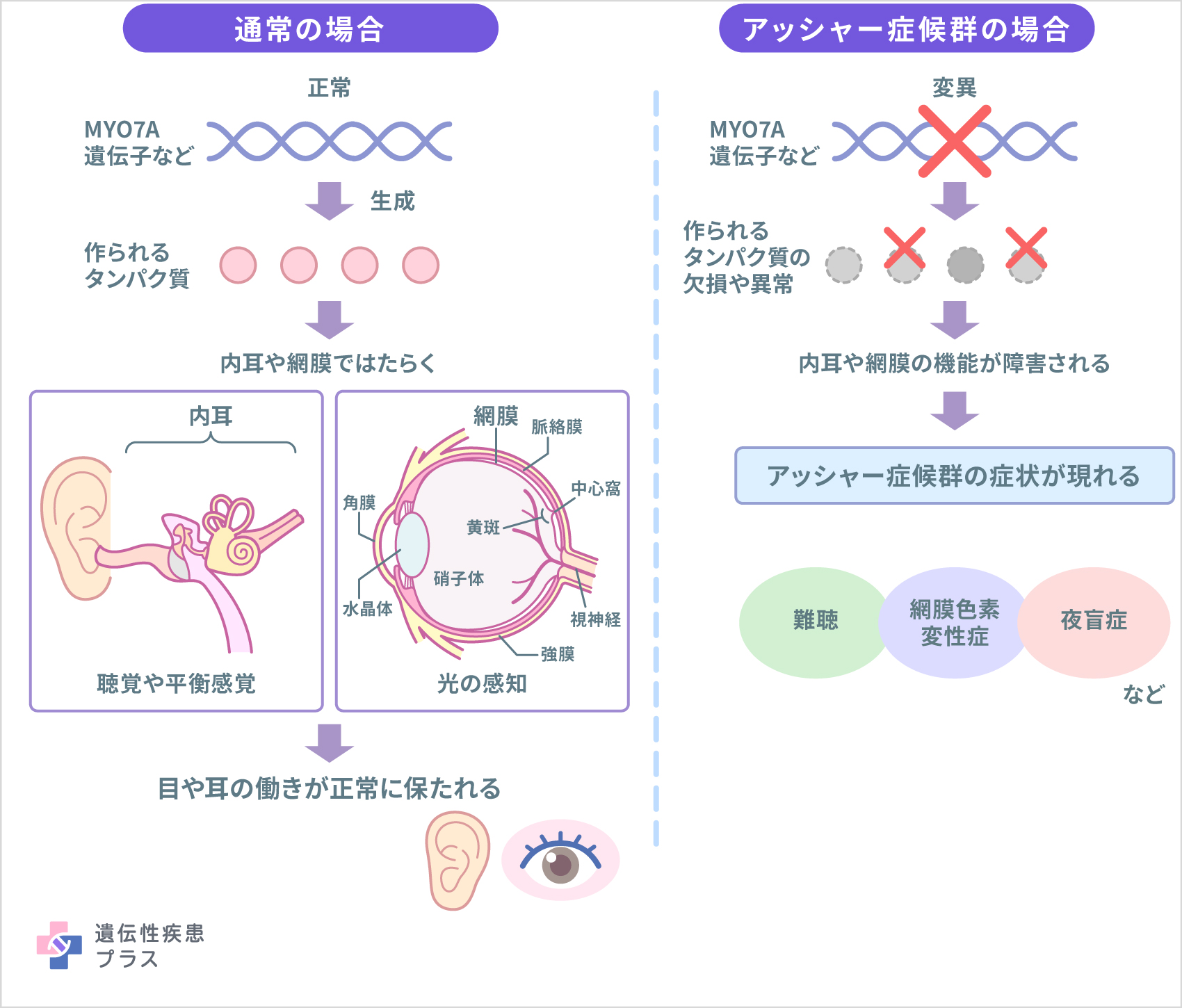
アッシャー症候群の原因遺伝子として現在までに10種類以上が見つかっており、タイプ1に関わる遺伝子はMYO7A、USH1C、CDH23、PCDH15、USH1G、CIB2、タイプ2はUSH2A、ADGRV1(GPR98)、WHRN(DFNB31)、タイプ3はCLRN1です。また、海外の報告ではタイプ3の遺伝子としてHARS1も報告されています。これらの遺伝子から産生されるタンパク質は、内耳においては聴覚や平衡感覚に関与する有毛細胞の機能、網膜では光感知細胞の維持に関与しており、これらの遺伝子の変異によって内耳および網膜の機能が障害されると考えられています。
アッシャー症候群は常染色体劣性(潜性)遺伝形式で遺伝します。両親がともにアッシャー症候群に関わるいずれかの遺伝子の片方に変異を持つ(保因者)場合、子どもは4分の1の確率でアッシャー症候群を発症します。また、2分の1の確率で保因者となり、4分の1の確率でこの遺伝子の変異を持たずに生まれます。

どのように診断されるの?
国内のアッシャー症候群に関する調査研究班によって以下の診断基準が作成されています。
1.自覚症状
A.夜盲、視野狭窄、視力低下などの視覚障害(網膜色素変性症)
B.両側性感音難聴、めまいなどの耳症状(蝸牛・前庭症状)
2.臨床検査所見
A.網膜色素変性症に関する所見(以下のうち、網膜電位の異常を含めて2つ以上を満たす)
(1) 眼底所見:網膜血管狭小、粗造胡麻塩状網膜、骨小体様色素沈着、多発する白点など
(2) 網膜電位の異常(振幅低下、または消失)
(3) 蛍光眼底造影所見で網膜色素上皮萎縮による過蛍光または低蛍光
(4) 光干渉断層像で中心窩におけるIS/OSの異常(不連続または消失)
B.感音難聴に関する所見(以下の全てを満たす)
(1) 純音聴力閾値検査(気導・骨導)の閾値上昇
(2) 中枢性疾患、Auditory Neuropathy、伝音難聴が否定できる
3.疾患のタイプ分類
タイプ1:先天性の高度~重度難聴を呈する。両側前庭機能障害を伴う例が多く、視覚症状は10歳前後より生じる。
タイプ2:先天性の高音障害型の難聴を呈する。視覚症状は思春期以降に生じる。前庭機能は正常である例が多い。
タイプ3:難聴、視覚症状とも思春期以降に生じ、難聴は徐々に進行。
4.遺伝学的検査
原因遺伝子としては現在までに10遺伝子が同定されている。タイプ1はMYO7A、USH1C、CDH23、PCDH15、USH1G、CIB2であり、タイプ2はUSH2A、ADGRV1(GPR98)、WHRN(DFNB31)、タイプ3はCLRN1である。
<診断のカテゴリー>
「1-Aと2-A」および「1-Bと2-B」の双方を満たす場合、もしくは「1-Aと2-A」または「1-Bと2-B」のいずれかを満たし、4.遺伝学的検査により特異的な遺伝子変異を認める場合にアッシャー症候群と診断する。
どのような治療が行われるの?
アッシャー症候群自体に対する根治的治療はなく、聴覚や視覚に対する治療が行われます。難聴に対する治療は、難聴の程度に応じて、補聴器や人工内耳によって聴力を補う治療が行われます。特に先天性の高度難聴を呈するタイプ1に対しては補聴器の効果は限定的であることから、人工内耳の装用も考慮されます。補聴器や人工内耳によって聴覚刺激によって聴覚・言語に発達を促すことが重要になります。また、将来的に網膜色素変性症が進行し、社会的失明となる可能性を考慮し、早期からの両側人工内耳装用により聴覚情報を担保することが予後のために重要であるとされています。
網膜色素変性症に対する治療はその進行を抑えることを目的として、ビタミンA、循環改善薬などの内服薬が用いられます。また、遮光眼鏡の使用が暗順応障害(暗順応とは明るい状態から暗い状態になった場合に暗さに目が慣れること)や進行予防に有効との報告もありますが、その有効性は十分に証明されていません。米国の遺伝性疾患情報サイト「GeneReviews」では、視覚障害を想定したコミュニケーション手段として手話、読唇術、触手話(触手話は手話の形を触って読み取る方法)も考慮する旨が示されています。
どこで検査や治療が受けられるの?
日本でアッシャー症候群の診療を行っていることを公開している、主な施設は以下です。
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
難病の患者さん・ご家族、支えるさまざまな立場の方々とのネットワークづくりを行っている団体は、以下です。
参考サイト
- 難病情報センター
- MedlinePlus
- Genetic and Rare Diseases Information Center
- Online Mendelian Inheritance in Man(R) (OMIM(R))
- orphanet
- GeneReviews






