



どのような病気?
フォンヒッペル・リンドウ病は、多臓器に腫瘍や嚢胞ができ、大きくなったり再発したりする、遺伝性疾患です。発生する主な腫瘍・嚢胞は、中枢神経系(小脳、脊髄、脳幹)の血管芽腫、網膜の血管腫、腎細胞がん、副腎の褐色細胞腫、膵病変(膵嚢胞、膵ラ氏島腫瘍)、精巣上体嚢胞、側頭骨の内リンパ管腺腫などで、体のどちらか一方に偏ることなく、両側に発生します。これらの腫瘍は、生涯にわたり多発・再発します。腫瘍は通常良性ですが、腎臓や膵臓にできる一部の腫瘍は、他の臓器に転移して命に関わることがあります。
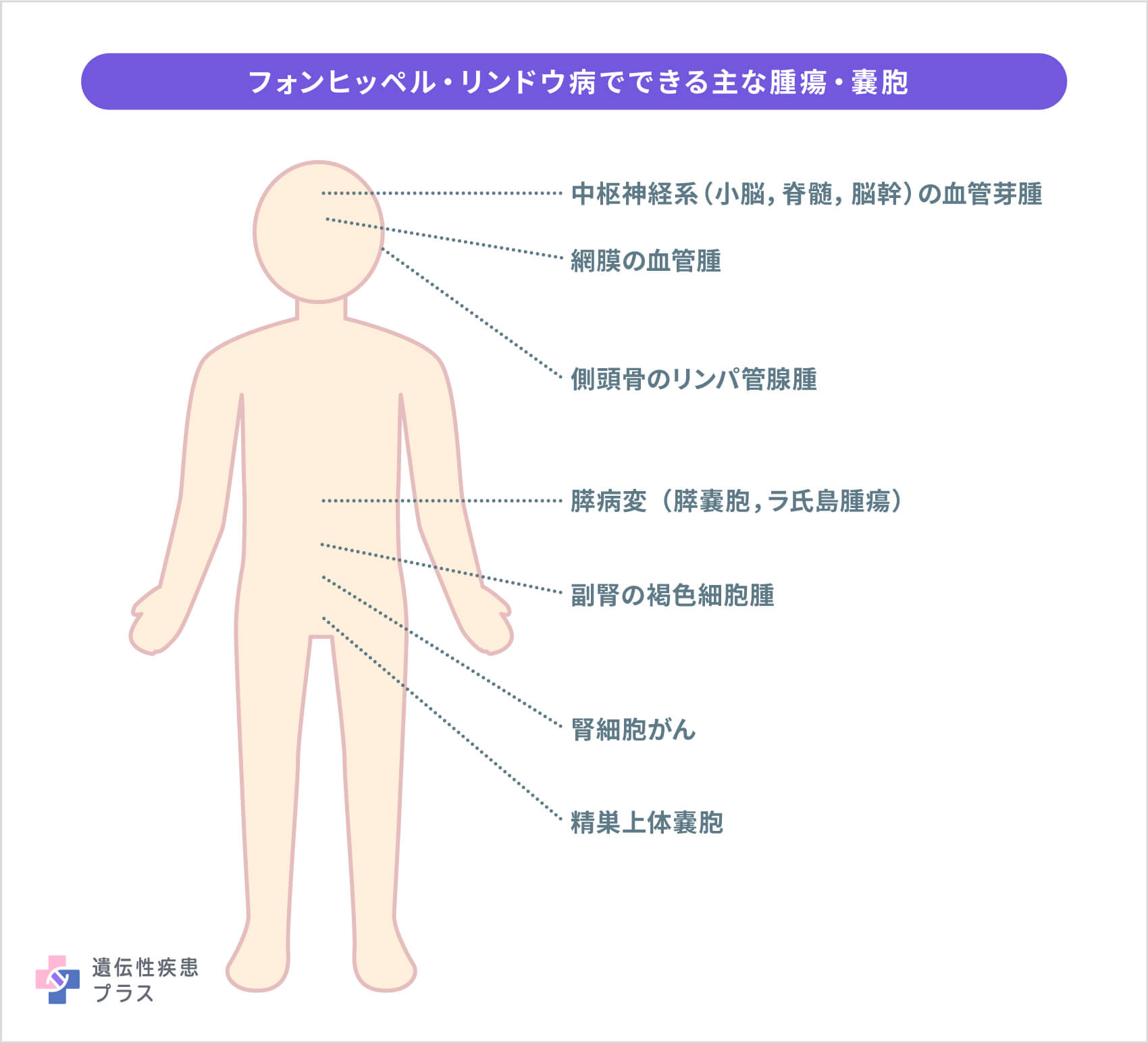
多発・再発する腫瘍の合併症は、腫瘍の大きさと場所によって異なりますが、頭痛、歩行失調、めまい、手足の脱力、視力低下、高血圧などです。また、頻回の腫瘍摘出手術が原因で、生活の質に関わる身体的な障害を合併する人も多くいます。例えば、網膜の手術による視力障害、脳脊髄の手術による手足のまひ、腎臓の手術後の腎機能不全、精巣上体腫瘍摘出後の不妊症などです。
| フォンヒッペル・リンドウ病でみられる症状 |
|---|
79%~30%でみられる症状 副腎褐色細胞腫、小脳血管芽腫、尿中カテコールアミンの上昇、腎細胞がん、網膜毛細血管腫 |
29%~5%でみられる症状 腹痛、左心室機能の異常、不安、背中の痛み、心筋症、遠位下肢筋力低下、血中カテコールアミンの上昇、内リンパ嚢腫瘍、頭痛、多汗症、高血圧性網膜症、手足の痛み、黄斑浮腫、多発性腎嚢胞、肌の蒼白、動悸、膵嚢胞、膵ラ氏島細胞腺腫、精巣上体の乳頭状嚢胞腺腫、うっ血乳頭、脳卒中、上肢の筋力低下、めまい、視力喪失 |
4%~1%でみられる症状 精巣上体嚢胞、頭蓋内圧の上昇、心筋梗塞、心筋炎、真性赤血球増加症(多血症)、網膜剥離 |
割合は示されていないがみられる症状 肝臓の異常、膵臓がん、褐色細胞腫、肺毛細血管腫、感音難聴、脊髄血管芽腫、耳鳴り |
腫瘍が最初に現れる年齢は3、4歳~70歳代と広範囲ですが、多くの場合、若年成人期(10~30歳代)に最初に現れます。網膜血管腫は10歳前後、中枢神経系血管腫は20歳前後、褐色細胞腫は20~30歳、腎細胞がんは30歳代後半に多く発症します。
小児慢性特定疾病情報センターのウェブサイトに掲載の情報(2014年)によると、日本にこの病気の家系は200家系程度、患者さんは600~1,000人存在すると推定されています。発症頻度は、欧米では3万6,000人に1人程度とされています。
フォンヒッペル・リンドウ病は、18歳未満の場合は小児慢性特定疾患治療研究事業の対象疾患に指定されており、公費対象となっています。
何の遺伝子が原因となるの?

フォンヒッペル・リンドウ病の原因遺伝子は、3番染色体の3p25.3という位置に存在する「VHL遺伝子」であるとわかっています。この遺伝子はがん抑制遺伝子の仲間で、「VHLタンパク質」の設計図となる遺伝子です。
VHLタンパク質は、「VCB-CUL2複合体」を構成する複数のタンパク質のうちの1つです。VCB-CUL2複合体は、「ユビキチンープロテアソーム系」という仕組みにより細胞内で不要になったタンパク質を分解し、細胞の正常な機能を維持するために働いていますが、その標的の1つに「HIF-2α」と呼ばれるタンパク質があります。HIF-2αはHIFと呼ばれるより大きなタンパク質複合体の一部(サブユニット)です。HIFは、酸素の少ない環境では、赤血球を増やしたり、新しい血管を形成したりするVEGF遺伝子などを活発に働かせ、低酸素に対応します。VHLタンパク質に異常があり、VCB-CUL2複合体が正常に働かないと、十分な酸素が利用できる環境下でもHIFが細胞内に不適切に蓄積し、VEGF遺伝子などが過剰に働くことで腫瘍が生じます。これが、フォンヒッペル・リンドウ病の原因の1つであると考えられていますが、VHL遺伝子の異常と、さまざまな部位に発生する腫瘍との関係は、まだ完全には解明されていません。
それ以外にも、VHLタンパク質は、複数の遺伝子の調節を通じて細胞分裂を適切に制御し、がん抑制因子として働いている可能性が考えられています。また、VHLタンパク質は、組織の構造を保つために重要な「細胞外マトリックス」の形成にも関与していることがわかってきています。
フォンヒッペル・リンドウ病でみられるVHL遺伝子の異常は、約6割がミスセンス変異(作られるタンパク質のアミノ酸が変化し、機能不全になる)、約4割がフレームシフト(タンパク質の設計図としての遺伝子の読み枠がずれて機能的なタンパク質が作れなくなる)や、ナンセンス変異(変異した部分以降がない不完全なタンパク質となる)で、フレームシフトやナンセンス変異は腎細胞がんと関連するとされています。
フォンヒッペル・リンドウ病は、常染色体優性(顕性)遺伝形式で親から子へ遺伝するため、患者さんが子どもを持った場合、親から子への遺伝は50%(2人に1人)の確率であり得ます。親から遺伝するのではなく、子どもに新たな変異が起こり発症する例もあります(このような発症パターンを孤発例と言います)。親からの遺伝で発症する人(家族性)は8~9割、孤発例は1~2割とされています。
常染色体優性(顕性)遺伝するほとんどの疾患は、両親から1コピーずつ計2コピー受け継いだ遺伝子のうち、変異した原因遺伝子が1コピーあることが原因で発症します。しかし、フォンヒッペル・リンドウ病は、変異したVHL遺伝子を1コピー受け継いで生まれた後、生きていく過程でもう1コピーのVHL遺伝子にも変異を生じ、フォンヒッペル・リンドウ病を発症します。親から変異のある遺伝子を受け継いだ場合に実際に発症する確率を「浸透率」と言いますが、フォンヒッペル・リンドウ病は浸透率がほぼ100%と知られています。
どのように診断されるの?
日本小児神経学会による診断の手引きには、「フォンヒッペル・リンドウ病の診断方法」として以下が記載されており、これらの症状や所見から、フォンヒッペル・リンドウ病の診断が行われます。
フォンヒッペル・リンドウ病の家族歴がある場合
中枢神経系(小脳、脊髄、脳幹)の血管芽腫、網膜血管腫、腎細胞がん、腎嚢胞(多発性)、褐色細胞腫、精巣上体嚢胞、膵嚢胞(多発性)のどれかがあるとき、フォンヒッペル・リンドウ病と診断されます。
フォンヒッペル・リンドウ病の家族歴がない場合
「中枢神経系(小脳、脊髄、脳幹)の血管芽腫または網膜血管腫が2つ以上ある」または「網膜血管腫があり、腎細胞がん、腎嚢胞(多発性)、褐色細胞腫、精巣上体嚢胞、膵嚢胞(多発性)、膵ラ氏島腫瘍、内リンパ管腺腫(側頭骨内)のどれかがある」とき、フォンヒッペル・リンドウ病と診断されます。
重要な検査所見として、
- VHL遺伝子変異(未発症患者を含め、確定診断に有用)
- 高血圧
- 多血症(ヘモグロビン(Hb)値 16.0g/dl以上)
が挙げられています。
どのような治療が行われるの?
今のところ、フォンヒッペル・リンドウ病を根本的に、つまり、遺伝子から治すような治療法は見つかっていません。
そのため、発生した腫瘍の早期発見が重要で、全ての腫瘍に対して摘出手術、場合によっては罹患臓器の摘出手術による治療が行われています。
網膜の血管腫はレーザー焼灼術で治療が行われることもあります。脳腫瘍(中枢神経系の血管芽腫)に対しては、定位放射線治療(ガンマナイフ治療)が行われることもありますが、まだ十分な検証は行われていません。褐色細胞腫に対しては、腹腔鏡手術も試みられています。
海外では、HIF-2αの働きを阻害する薬剤について、フォンヒッペル・リンドウ病による腫瘍発生を抑える薬としての臨床試験が進められています。
どこで検査や治療が受けられるの?
日本でフォンヒッペル・リンドウ病の診療を行っていることを公開している、主な施設は以下です。
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
フォンヒッペル・リンドウ病の患者会で、ホームページを公開しているところは、以下です。
参考サイト
- 難病情報センター
- 小児慢性特定疾病情報センター フォンヒッペル・リンドウ(von Hippel-Lindau)病
- MedlinePlus
- Genetic and Rare Diseases Information Center
- Online Mendelian Inheritance in Man(R) (OMIM(R))
- MD Anderson Newsroom
- 遺伝性疾患プラス 「遺伝性のがん」ってどんながん?―子どもへの遺伝から検査・予防まで






