



どのような病気?
ワーデンブルグ症候群(WS)は、先天性の感音難聴、目の虹彩の色素異常(淡く青い色の目、片方が青で片方が茶色など両目の色が異なる、1つの目に2色含まれている、など)、毛髪の色素異常(白い前髪、30歳前に頭髪が白髪になる、など)、皮膚の色素異常(先天性白斑など)を特徴とする、希少な遺伝性疾患です。
症状は人によってさまざまで、同じ家族でも異なる場合があります。
| ワーデンブルグ症候群でみられる症状 |
|---|
99%~80%でみられる症状 顔の形の異常、視力の異常、伝音難聴、異なる色の目、皮膚の白斑(部分白子症)、頭髪の若白髪、高い鼻梁(びりょう、鼻筋のこと)、眉毛叢生(両側の眉毛がくっついて生えている) |
79%~30%でみられる症状 唇の形の異常、涙の分泌異常、眼角開離(目の角と角が大きく離れている)、鼻孔の形成不全、白い前髪、広い鼻梁 |
29%~5%でみられる症状 膣の形態異常、子宮の奇形、無神経節性巨大結腸症(神経細胞が欠落しており肥大した結腸)、結腸の欠如・未発達、腸閉塞、脊髄髄膜瘤、口唇裂、眼瞼下垂 |
割合は示されていないがみられる症状 膣形成不全、眼瞼裂狭小、青い目、先天性感音難聴、間隔の広い目、眼底の色素脱失、虹彩間質の発育不全、大きな下顎、部分的な白皮症、平坦な人中、二分脊椎、肩甲骨が高い、肋骨の数が多い、脊椎骨の数が多い、太い眉毛、白い眉毛、白いまつげ、精神運動発達遅滞 |
WSは、症状や遺伝学的な特徴から4つの型(WS1~WS4)に分類されます。WS1とWS2は、特徴がよく似ていますが、WS1はほとんどの場合で眼角開離が認められ、WS2では眼角開離が認められません。また、難聴は、WS1よりWS2の人で多く発症します。WS3は、「クライン・ワーデンブルグ症候群」と呼ばれることもあります。WS3もWS1に似ていますが、腕や手の関節がまっすぐにならない、指がくっついている、あるいは水かきがあるなどの筋骨格異常がみられます。WS4は、「ワーデンブルグ・シャー症候群」と呼ばれることもあります。WS4はWS2の症状に加え、ヒルシュスプルング病という、重度の便秘や腸閉塞を引き起こす腸障害の所見が認められるという特徴があります。
WSの発症率は、全世界では4万人に1人程度、日本では5万人に1人程度と推定されています。中でも、WS1とWS2が多く、WS3とWS4はまれです。特に、WS3は、まだ世界で数例しか報告がありません。また、先天性難聴患者さん全体の2~4%がWSだという報告もありますが、実態を明確に示すための大規模な疫学調査データはまだありません。
何の遺伝子が原因となるの?
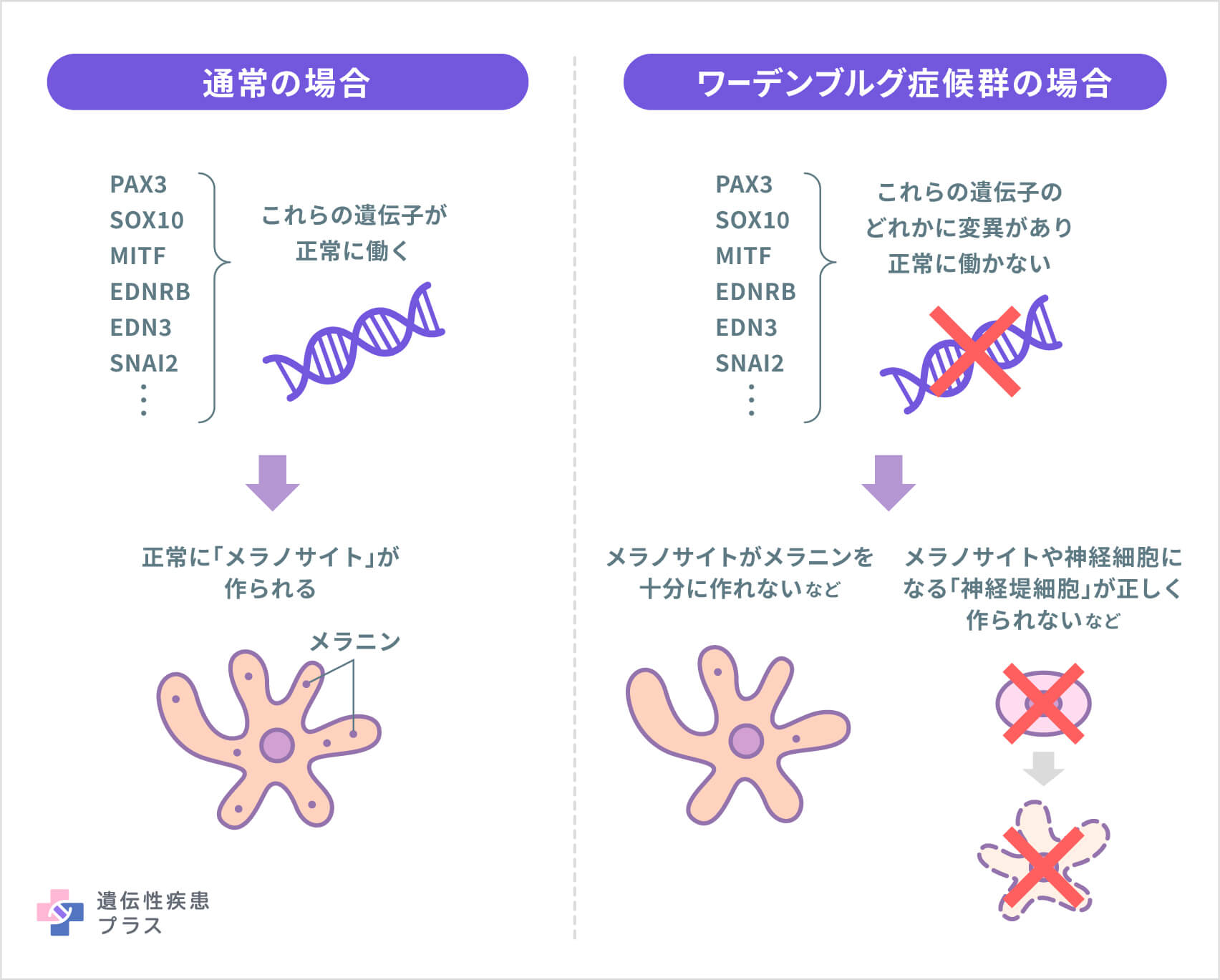
WSの原因となる遺伝子は複数あり、今のところ、変異が見つかっている遺伝子は、PAX3、SOX10、MITF、EDNRB、EDN3、SNAI2の6つです。これら6つの遺伝子に変異が見られず、まだ原因遺伝子が特定されていないWSの人もいます。
これら6つの遺伝子は、「メラノサイト」と呼ばれる色素産生細胞を含む、数種類の細胞の形成や成長に関与しています。メラノサイトは「メラニン」と呼ばれる色素を作ります。メラニンは皮膚、髪、目の色に関わる一方で、内耳が正常に機能するために不可欠な役割も持っています。そのため、これら6つの遺伝子のどれかに変異があると、メラノサイトが正常に作られず、皮膚、髪、目の色が異常になったり、聴覚に問題が起きたりするといった、WSの症状が現れます。
WSで変異が見つかっている6つの遺伝子
| 遺伝子 | 染色体位置 | 作られるタンパク質 | 主な役割 | 変異が見られる病型 |
|---|---|---|---|---|
| PAX3 | 2q36.1 | ペアードボックス3(転写因子) | メラノサイトのもととなる神経堤細胞の形成 | WS1、WS3 |
| SOX10 | 22q13.1 | SRY-ボックス10(転写因子) | 腸管神経やメラノサイトの形成 | WS2、WS4 |
| MITF | 3p13 | メラノサイト誘導転写因子 | メラノサイトの成長や色素生成の制御 | WS2 |
| EDNRB | 13q22.3 | エンドセリン受容体B型 | 腸管神経やメラノサイトの形成 | WS4 |
| EDN3 | 20q13.32 | エンドセリン3 | 腸管神経やメラノサイトの形成 | WS4 |
| SNAI2 | 8q11.21 | スネイルファミリー転写抑制因子2 | メラノサイトのもととなる神経堤細胞の形成 | WS2 |
この病気は、通常、常染色体優性(顕性)遺伝と呼ばれる形式で遺伝します。つまり、両親のどちらかがWSだった場合、子どもがWSになる確率は50%だといえます。ただし、全員が親からの遺伝でこの病気になっているわけではありません。一部の患者さんは、両親ともにWSではなく、出生時に遺伝子が新たに変異することでこの病気になっています。

また、WS2とWS4の一部の患者さんでは、2つセットでもつ遺伝子の両方に変異が確認されており、常染色体劣性(潜性)遺伝と呼ばれる形式で遺伝する場合もあると考えられています。この遺伝形式の場合、2つセットで持っている原因遺伝子のうち、両親がともに1本ずつ変異を有していた場合、子どもは4分の1の確率で2本とも変異を有してWSを発症します。

どのように診断されるの?
日本には、まだWSの診断基準が設けられておらず、その人の症状や家族歴、遺伝子変異などの情報をもとにして、医師が診断をしています。一方で、いま、遺伝子診断を用いた新しい診断法を日本で確立し、遺伝子診断に基づいた診断基準を確立するための準備が進められています。
信州大学を中心とした研究グループは、2017年から2018年にかけ、国の研究課題として全国80施設の共同研究施設を対象に、「ワーデンブルグ症候群の診断基準および重症度分類策定に関する調査研究」を行いました。さまざまな臨床症状がとりまとめられ、遺伝子解析の結果と合わせて解析が行われた結果、診断基準(案)が作成されました。その際、6つの主な遺伝子変異に加え、新たに13の遺伝子変異も見つかっています。この診断基準(案)では、大症状1つに加え、遺伝子変異が認められた場合、WSと確定診断されます。
国際的には、米国が中心となったワーデンブルグコンソーシアムが1992年に示した診断基準があります。メジャー基準を「先天性感音難聴、虹彩異色症、白い前髪など頭髪の色素異常、眼角開離、1親等にWSの患者さんがいる」、マイナー基準を「出生時からの皮膚の白斑、眉毛叢生、高い鼻梁、鼻孔の形成不全、30歳前で頭髪の若白髪」とし、メジャー基準が2つ以上、もしくはメジャー基準が1つとマイナー基準が2つ以上当てはまる場合、WS1と診断されます。WS1と同様の基準で眼角開離が認められない場合、SW2と診断されます。また、WS1に加え、腕や手の関節がまっすぐにならない、指がくっついている、あるいは水かきがあるなどの筋骨格異常がみられる場合、WS3と診断されます。WS2に加え、ヒルシュスプルング病の所見が認められる場合、WS4と診断されます。
どのような治療が行われるの?
今のところ、WSを根本的に、つまり、遺伝子から治すような治療法は見つかっていません。そのため、難聴には補聴器や人工内耳を用いるなど、それぞれの症状に合わせた対症療法が行われます。
どこで検査や治療が受けられるの?
日本でワーデンブルグ症候群の診療を行っていることを公開している、主な施設は以下です。
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
難病の患者さん・ご家族、支えるさまざまな立場の方々とのネットワークづくりを行っている団体は、以下です。
参考サイト
- KEGG DISEASE:ワールデンブルグ症候群
- 厚生労働科研研究成果データベース ワーデンブルグ症候群の診断基準および重症度分類策定に関する調査研究
- MedlinePlus
- Genetic and Rare Diseases Information Center
- Orphanet






