



どのような病気?
コケイン症候群は、日光に対する感受性が高い、精神運動発達遅滞、腎不全、難聴、視力障害、歩行障害などのさまざまな症状が見られるまれな遺伝性疾患です。1936年にイギリスの小児科医、コケイン博士により最初に報告されたことからこの名前が付けられました。
この病気の症状が見られ始める時期は人によってさまざまですが、多くは幼少期から始まり、時間の経過とともに症状が進行します。多くの場合、生まれた時から日に当たると赤くなりやすく、日光の感受性が高い(光線過敏症)症状が見られ、場合によっては少しの日光を浴びただけでも日焼けや皮膚の水ぶくれを引き起こすことがあります。また、発育や発達の遅延が見られ、年齢や月齢で期待される速度での体重や身長の成長が見られず、それに伴う低身長や小頭症(異常に小さい頭)なども特徴となります。
その他の症状として、特有の早老様の顔立ち(小頭、目のくぼみ、皮下脂肪萎縮など)、難聴(感音性難聴)、視力障害(視力低下、網膜色素変性)、重度の虫歯など歯の異常、手足が冷たい、脳のスキャンで見られる脳の変化、などがあります。また、この病気の人は「メトロニダゾール」と呼ばれる抗生物質に対して異常に反応することがあり、コケイン症候群の人がこの薬を服用すると、肝不全を引き起こして命に関わることもあるとされています。またこの病気は、色素性乾皮症(XP)と合併する場合もあるとされています。
コケイン症候群は、症状の重症度や症状が見られ始めた年齢によって、いくつかのタイプ(I型~III型、COFS症候群)に分けられることがあり、I型は、古典的なコケイン症候群の症状かつ中等度の病型、II型は、出生時から症状が見られるより重篤な形態(脳眼筋骨格症候群とも呼ばれる)、III型は、より軽度で発症が遅い病型とされています。以下にそれぞれのタイプで定義されている症状や特徴を示します。
I型は、出生前の成長は正常ですが、生後2年までに成長や発達に遅れが現れ始めます。病気の症状がはっきり見られる頃には、身長、体重、頭囲は全体の5%を下回っているとされます。また、視力、聴力のほか、神経系の機能障害が進行し、重度の障害を引き起こします。15歳前後からは腎障害、肝機能障害、心血管障害、高血糖、呼吸器系・尿路系感染症などの合併にも注意が必要になります。10~20代で命を落とすこともあります。国内で見られるコケイン症候群のほとんどはI型とされています。
II型は、出生時の成長不全を特徴とし、出生後から神経の発達はほとんどありません。先天性白内障や眼の構造異常などが存在する場合もあります。この病型では、出生後早期に脊椎の拘縮(脊柱後弯症、側弯症)や関節の拘縮が見られます。通常、5歳までに亡くなることが多い病型とされています。
III型は、この病気に関連する主要な症状や特徴が2歳以降になってから明らかになる病型で、成長や認知能力はI型よりも良好とされています。
しかし、タイプ間の違いは必ずしも明確ではなく、症状は連続したスペクトルを示していると考える専門家もいます。
コケイン症候群で見られる症状 |
|---|
高頻度に見られる症状 進行性小頭症、重度の低身長、小脳萎縮、脳髄鞘形成不全(髄鞘は神経細胞の周りの膜構造、ミエリンとも呼ばれる)、網膜色素異常、色素性網膜症、悪液質(慢性疾患に関連する全身の衰弱)、成長遅延、出生後の成長遅延、認知機能低下、進行性感音難聴、行動異常 |
良く見られる症状 乾燥した髪、細い髪、深い眼球(顔の平面に対し目が通常より深くへこんでいる)、眼の形態異常、早期の皮膚のしわ、基底核石灰化(脳内の大脳基底核と呼ばれる領域のカルシウム沈着)、脳萎縮、神経膠症(しんけいこうしょう、グリオーシス)、皮質下白質の斑状脱髄、脳石灰化、皮質下白質石灰化、小脳歯状核の高密度石灰化、白内障、網膜ジストロフィー、進行性の視力低下、高周波感音難聴、歯のエナメル質形成不全、虫歯、皮膚の光線過敏症、骨格筋の萎縮、先天性関節拘縮、大関節の拘縮、皮下脂肪組織の減少、胃食道逆流症、乳児期の摂食障害、脱髄性末梢神経障害、感覚運動神経障害、末梢神経障害、感覚障害、小脳性運動失調症、歩行障害、進行性歩行失調症、てんかん発作認知障害、全般的な発達遅滞 |
しばしば見られる症状 先天性小頭症、凸型の鼻堤、頬部発疹、頭蓋骨の肥厚、球形円錐水晶体(眼の水晶体の湾曲が過剰になり球状に膨らむ)、瞳孔収縮、網膜変性、網膜細動脈収縮、角膜の形態異常、網膜電図(網膜の電気的反応)の異常、帯状角膜変性、角膜潰瘍、乾性の角結膜炎、先天性白内障、斜視、遠視、眼振、視神経萎縮、視神経乳頭の蒼白、無涙症(涙液が分泌されない)、涙液分泌の減少、乳歯の萌出遅延、歯の数の異常、歯の形態異常、歯の不正咬合、アテローム性動脈硬化症、血管石灰化、高血圧、肝腫大、脾腫、片側腎無形成症、腎低形成、腎不全、腎機能異常、ネフローゼ症候群、停留精巣、神経損傷による膀胱機能障害、尿失禁、骨端の形態異常、脊柱後弯症、脊柱側弯症、猫背の姿勢、光過敏症、乳児期の胃ろうチューブ栄養が必要な摂食障害、無汗症、末梢軸索神経障害、痙縮(けいしゅく)、動作時振戦、意図振戦、反射亢進、反射消失、反射低下、体軸性筋緊張低下、筋緊張亢進、四肢の筋緊張亢進、歩行困難、歩行不能、弱々しい泣き声、思春期の発達の欠如、思春期の遅れ、発達退行、知的障害 |
まれに見られる症状 小眼球症、永久歯の欠損、網膜萎縮、網膜出血、言語発達の欠如 |
コケイン症候群の発症頻度は50万人に1人とされ、非常にまれな病気です。日本における患者数は約50人とされています。
コケイン症候群は、指定難病対象疾病(指定難病192)および、コケイン(Cockayne)症候群として小児慢性特定疾病の対象疾患となっています。
何の遺伝子が原因となるの?
コケイン症候群は、10番染色体の10q11.23領域に存在するERCC6遺伝子、5番染色体の5q12.1領域に存在するERCC8遺伝子の変異によって起こることが知られています(ERCC6遺伝子はCSBとも呼ばれ、ERCC8遺伝子はCSAとも呼ばれます)。ERCC6の病原性変異は、この病気の人の約65%、ERCC8の病原性変異は約35%を占めるとされています。また、色素性乾皮症の原因であるXPB(2q14.3)、XPD(19q13.32)、XPG(13q33.1)遺伝子が関与することもあるとされます。
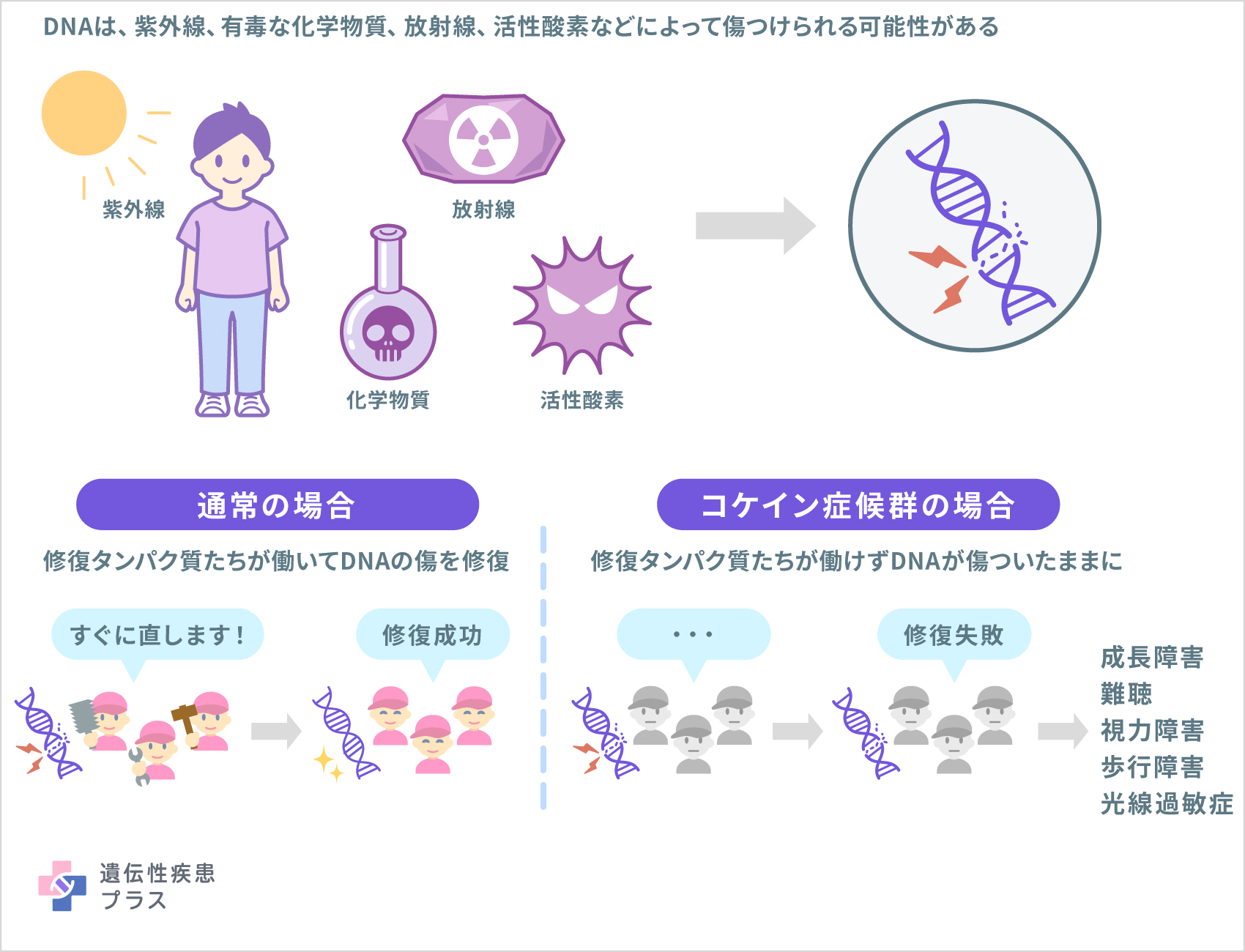
これらの遺伝子は、何らかの理由で損傷したDNAの修復に関与するタンパク質の設計図となります。DNAは、太陽からの紫外線や有毒な化学物質、放射線、活性酸素などによって、常に傷つけられる可能性があります。通常、細胞は何らかの問題が起こる前にDNAの損傷を修復します。しかし、コケイン症候群の場合には、ERCC6遺伝子やERCC8遺伝子の変異によってDNAが正常に修復されません。このようなDNAの傷(エラー)が蓄積することで、細胞は機能不全に陥り死滅します。
このようなDNA修復の機能不全は、この病気の光線過敏症と関連しており、コケイン症候の他の特徴にも関与している可能性があります。その一方でERCC6遺伝子やERCC8遺伝子の変異が、この病気のさまざまな症状のすべてをどのように引き起こすのかについては、まだわかっていません。
コケイン症候群は、常染色体劣性(潜性)遺伝形式で遺伝し、両親から受け継いだ2つの原因遺伝子に変異が生じることで発症します。2つの遺伝子のうち、1つに変異がありもう一方には変異がなく機能を持っている場合には、病気の保因者となりますが、通常、症状や特徴は現れません。両親が2人ともこの病気の保因者である場合に、コケイン症候群の子どもが生まれる可能性は25%です。

どのように診断されるの?
コケイン症候群の診断基準について、難病情報センターのwebサイトには以下のように記載されています。
コケイン症候群の各種症状
主徴候
(1)著明な成長障害
2歳で身長、体重、頭囲が5パーセンタイル以下。
2歳以降はさらにパーセンタイル値が減少する。I型、XP合併型では生後1歳前後から、II型では出生時から確認できるが、III型では成人期以降に出現あるいは見られないこともある。
(2)精神運動発達遅滞
言葉や歩行の発達が極めて遅いなどで気づかれる。
(3)早老様の特徴的な顔立ち(くぼんだ眼と頬、鳥のくちばし様の鼻など)
2歳前後で傾向が始まる。III型では見られない場合あり。
(4)日光過敏症状
臨床像はサンバーン様の紅斑、浮腫、水疱形成。
既往歴含む、思春期以降は軽減傾向あり。
副徴候
乳児期にはまれ、幼児期以降に始まることが多い、(5)(6)は典型例で2歳前後までにはで確認できるが、(7)は年長になって出現することが多い。
(5)大脳基底核石灰化
(6)感音性難聴
(7)網膜色素変性症
その他の徴候(年齢とともに出現、進行するが、コケイン症候群に対する特異性は低い)
(8)白内障(II型では生下時から)
(9)足関節拘縮(II型では生下時から)
(10)視神経萎縮(II型では生下時から)
(11)脊椎後弯
(12)齲歯
(13)手足の冷感
(14)性腺機能低下
(15)睡眠障害
(16)肝機能障害
(17)耐糖能異常
予後に影響する合併症
(18)腎機能障害
(19)呼吸器感染
(20)外傷
(21)心血管障害
主徴候(1)~(4)のうち2項目以上があれば、以下のように診断が検討されます。
遺伝学的検査を行った結果、コケイン症候群関連遺伝子CSA、CSB、XPB(2q14.3)、XPD(19q13.32)、XPG(13q33.1)に病的変異が認められた場合にはこの病気の診断が確定となります。
遺伝学的検査で病的変異が未確定もしくは遺伝学的検査を未実施の場合には、以下a~cに従って診断が行われます。
a.主徴候(1)~(4)のうち2項目以上あり、紫外線感受性試験、宿主細胞回復を指標にしたDNA修復能測定、相補性試験、紫外線照射後RNA合成試験などのDNA修復試験での異常所見(修復能の低下があり、修復能の低下が既知のCS関連遺伝子導入で相補)を認めれば診断が確定となります。
b.主徴候(1)~(4)を全て満たし、DNA修復試験での異常所見(修復能の低下があり、修復能の低下が既知のCS関連遺伝子導入で相補しない、あるいは相補性試験未実施)を認めれば診断が確定となります。
c.DNA修復試験未実施の場合には、「主徴候(1)~(4)を全て、副徴候(5)~(7)のうち2項目以上」を満たし、「その他の臨床所見、血液・画像など各種データで他疾患(色素性乾皮症、ポルフィリン症など)が否定される」もしくは「同胞が同様の症状からCSと確定診断されている」があれば診断が確定となります。
どのような治療が行われるの?
コケイン症候群は、まだこの病気を根本的に治療する方法は確立されていません。そのため、この病気で見られる症状に対する対症療法が中心となります。
この病気の日光過敏症には、サングラス装着、紫外線防御クリームなど紫外線からの遮光、目や耳の症状には補聴器や眼鏡の使用などが勧められます。その他、栄養障害、感染症、腎障害、肝障害、糖尿病など症状ごとに治療が行われます。腎不全に対しては腹膜透析を行う場合もあります。
関節の拘縮や筋緊張に対してはリハビリも有用とされます。手指振戦に対してはTRH製剤と呼ばれるお薬が使われる場合もあります。
どこで検査や治療が受けられるの?
日本でコケイン症候群の診療を行っていることを公開している、主な施設は以下です。
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
コケイン症候群の患者会で、ホームページを公開しているところは、以下です。
参考サイト
- 難病情報センター
- 小児慢性特定疾病情報センター コケイン(Cockayne)症候群
- MedlinePlus
- Genetic and Rare Diseases Information Center
- Online Mendelian Inheritance in Man(R) (OMIM(R))
- KEGG DISEASE:コケイン症候群
- GeneReviews
- NORD






