



どのような病気?
先天性角化不全症は、皮膚や爪の異常をはじめ重度の貧血や肺の疾患など、さまざまな症状が引き起こされる遺伝性疾患です。先天性角化異常症と呼ばれる場合もあります。
この病気には典型的な3症状があり、それらは、1)手や足の爪の形が変形していたり成長が不良だったりすることなどの爪の異常、2)口の中に白い斑点ができる(口腔内白斑)、3)首や胸などの皮膚に見られるレース模様の色素沈着や皮膚萎縮、です。これら3症状の他にも、骨髄や肺など幅広く症状が見られます。
この病気の重い症状の一つとして、骨髄(血液細胞を作る臓器)の機能が低下することにより引き起こされる重度の貧血やがんがあります。骨髄は、血液細胞の元となる血球を産生していますが、骨髄の機能が失われることにより、十分な血球が作られず(血球減少)、重度の貧血(再生不良性貧血)に至る場合があります。また、骨髄で血球が十分に発達しないことによる骨髄異形成症のリスクが高くなります。骨髄異形成症は、白血病と呼ばれる血液のがんに進行する可能性があり、また先天性角化不全症の人は骨髄異形成症を発症していない場合でも白血病や、頭頸部、肛門、生殖器をはじめとするがんの発症リスクが平均よりも高いとされています。
肺で見られる症状の代表的なものとしては、肺線維症(肺の組織が硬くなって酸素を血液に送る機能が低下する)があります。それ以外の臓器や組織に見られる症状として、肝障害、骨格や骨の異常(股関節や肩関節の変性、骨密度低下など)、脱毛や白髪、目の異常、歯の異常などが見られるほか、男性において尿道が狭くなる尿道狭窄が発症し排尿困難や尿路感染症を引き起こす場合もあります。この病気では多くの場合、知能や運動能力は正常ですが、重症の患者さんの一部で発達遅延が見られる可能性もあります。
先天性角化不全症の重症度は、患者さんによって大きく異なり、軽度の障害や身体的特徴のみの場合もあれば、さまざまな症状をいくつも合併し、成人早期までに骨髄不全、がん、肺線維症のすべてを発症する場合もあります。
また、この病気には、Hoyeraal-Hreidarsson症候群、Revesz症候群、Coats plus症候群(石灰化と嚢細胞を伴う脳網膜微小血管症)と呼ばれる、以前は別の疾患として捉えられていた重症の疾患も含まれることがわかっています。Hoyeraal-Hreidarsson症候群は、先天性角化不全症の症状に加えて小脳が未発達で異常に小さく、子宮内発育遅延、発達遅延、小頭症、小脳形成不全、免疫不全などが認められます。
Revesz症候群では、重度の再生不良性貧血、子宮内発育遅延、細い毛髪、網状皮膚の色素沈着、小脳形成不全による運動失調、脳石灰化、進行性の精神運動発達遅滞のほか、両側性滲出性網膜症(目の中にある網膜の異常)を特徴とします。
また、Coats plus症候群は、爪の障害やまばらな髪などの典型的な皮膚粘膜の症状のほか、目、脳、骨、胃腸管などにも症状が見られ、両側性滲出性網膜症や網膜毛細血管拡張症、頭蓋内石灰化、骨減少症、消化管血管拡張症、発達遅延などが認められる場合があります。
先天性角化不全症で見られる症状 |
|---|
高頻度に見られる症状 爪の形態異常、爪ジストロフィー、皮膚の異常な水疱、色素沈着過剰の黒色斑、(皮膚の)しみ、口腔白板症、好中球の異常、貧血、血小板減少症 |
良く見られる症状 薄い頭髪、足の爪の形成不全、皮膚の形成不全、斑状に色素が欠失する、皮膚の毛細血管拡張症、皮膚の潰瘍、歯列の異常、虫歯、歯数の不足、歯周病、タウロドンティア(臼歯の形態異常、歯の頭の部分が長く歯根が短い)、咽頭の異常、食道狭窄症、気管食道瘻(きかんしょくどうろう、食道と気管の間に異常な通り道がある)、吸収不良(腸から栄養素を吸収する能力の低下)、尿道狭窄症、女性の内性器の異常、肛門と直腸の異常、骨幹端における粗い骨梁(骨を中から支える小さい柱)形成、血液凝固の異常、骨髄細胞減少、細胞性免疫と体液性免疫不全、呼吸器感染症を繰り返す、多汗症、新生物(腫瘍)、低身長、全般的発達遅滞、子宮内発育遅延、骨折を繰り返す |
しばしば見られる症状 眉毛の形態異常、まつげの形態異常、精巣の形態異常、脱毛症、無血管壊死(骨への血流が遮断され骨の組織が壊れる)、眼瞼炎(がんけんえん、まぶたの炎症)、白内障、脳内の石灰化、肝硬変、尿道口の位置の異常、聴覚障害、肝不全、肝腫大、上顎の形成不全、リンパ腫、膵臓腫瘍、骨粗しょう症、掌蹠角皮症、髪の早期白髪化、側弯症、皮膚小水疱(皮膚の小さな水ぶくれ)、脾腫、白髪 |
先天性角化不全症の正確な発症頻度や患者数は不明ですが、100万人に1人程度と推定されています。また、難病情報センターのウェブサイトには、日本における患者数は約200人(難治性疾患研究班情報・平成23年度)と記載されています。
先天性角化不全症は「原発性免疫不全症候群」として、国の指定難病対象疾病(指定難病65)に含まれる場合があります。また、「先天性角化異常症」として、小児慢性特定疾病の対象となっています。
何の遺伝子が原因となるの?
先天性角化不全症は、「テロメア」に関連した遺伝子の異常によって引き起こされることがわかっています。テロメアとは、それぞれの染色体の末端部分にあるDNAの繰り返し配列の領域で、染色体の構造が維持されるために重要な領域となっています。また、テロメアは細胞の寿命にも関わっています。ほとんどの細胞では、細胞が分裂し、染色体が複製されるたびにテロメアは少しずつ短くなります。細胞分裂がある一定の回数行われテロメアが非常に短くなると、その細胞は分裂できなくなり自己破壊します。先天性角化不全症の人は、テロメアが短かったり、非常に短くなったりします。
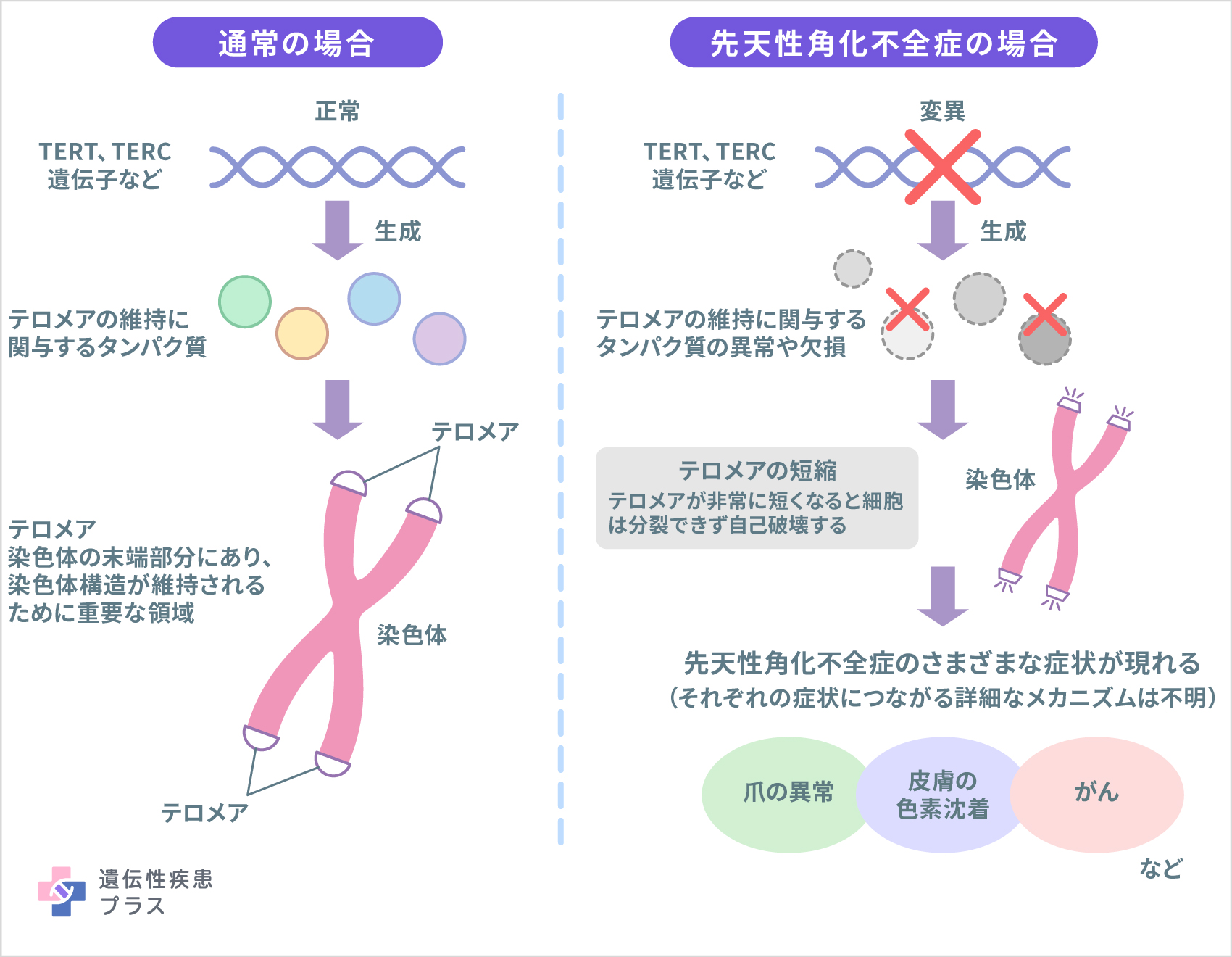
これまでに、先天性角化不全症の原因として複数の遺伝子が報告されており(下表参照)、これらの遺伝子はどれも異なる形でテロメアの維持に関わっています。
先天性角化不全症の原因遺伝子
原因遺伝子 | 染色体領域 | 作られるタンパク質など | 遺伝形式 |
|---|---|---|---|
TERT | 5p15.33 | テロメラーゼ逆転写酵素(hTERT) | |
TERC | 3q26.2 | テロメラーゼRNA構成要素(hTR) | 常染色体優性(顕性) |
DKC1 | Xq28 | ジスケリン(hTRに結合するタンパク質の一つ) | X連鎖劣性(潜性) |
TINF2 | 14q12 | シェルタリンタンパク質複合体因子TIN2(TERF1相互作用核因子2) | 常染色体優性(顕性) |
RTEL1 | 20q13.33 | テロメア伸長調節因子ヘリカーゼ1 | 常染色体劣性(潜性)または常染色体優性(顕性) |
ACD | 16q22.1 | シェルタリン複合体因子TPP1 | 常染色体劣性(潜性)または常染色体優性(顕性) |
NOP10 | 15q14 | NOP10リボ核タンパク質 | 常染色体劣性(潜性)または常染色体優性(顕性) |
WRAP53 | 17p13.1 | テロメラーゼカハール体タンパク質TCAB1 | 常染色体劣性(潜性) |
NHP2 | 5q35.3 | NHP2リボ核タンパク質 | 常染色体劣性(潜性) |
PARN | 16p13.12 | ポリ(A)特異的リボヌクレアーゼ | 常染色体劣性(潜性) |
CTC1 | 17p13.1 | CSTテロメア複製複合体 | 常染色体劣性(潜性) |
テロメアは、テロメラーゼとシェルタリンと呼ばれる2つのタンパク質複合体によって維持されています。正常なテロメアの長さを維持するために働くテロメラーゼは、TERC遺伝子を設計図とした「hTR」とTERT遺伝子を設計図とした「hTERT」で構成されます。また、DKC1遺伝子が設計図となるジスケリンは、hTRに結合し、テロメラーゼを安定化させる役割があります。
シェルタリンは、いくつかのタンパク質から構成されるタンパク質複合体で、DNAの修復機構のシステムが染色体の末端の構造を異常な切断として感知し、間違った修復や細胞の自己破壊を開始させてしまうことを防ぎます。TINF2遺伝子、ACD遺伝子はこのシェルタリン複合体を構成するタンパク質の設計図となります。
これらの遺伝子に変異が生じると、テロメラーゼやシェルタリンの機能不全を引き起こし、テロメアの維持障害やテロメア長の短縮につながる場合があります。特に、分裂回数の多い、爪や毛髪に関わる細胞、皮膚や口腔内の粘膜細胞、骨髄細胞などでは、テロメアが短縮する影響を受けやすくなると考えられ、先天性角化不全症で見られる症状が生じる可能性があります。しかし、それぞれの症状につながる詳細なメカニズムはまだわかっていません。
また、上記以外にも、TYMS、DCLRE1B、NAF1、POT1、RPA1、STN1、ZCCHC8遺伝子なども原因遺伝子として報告されているほか、まだ原因遺伝子がわかっていない先天性角化不全症もあります。
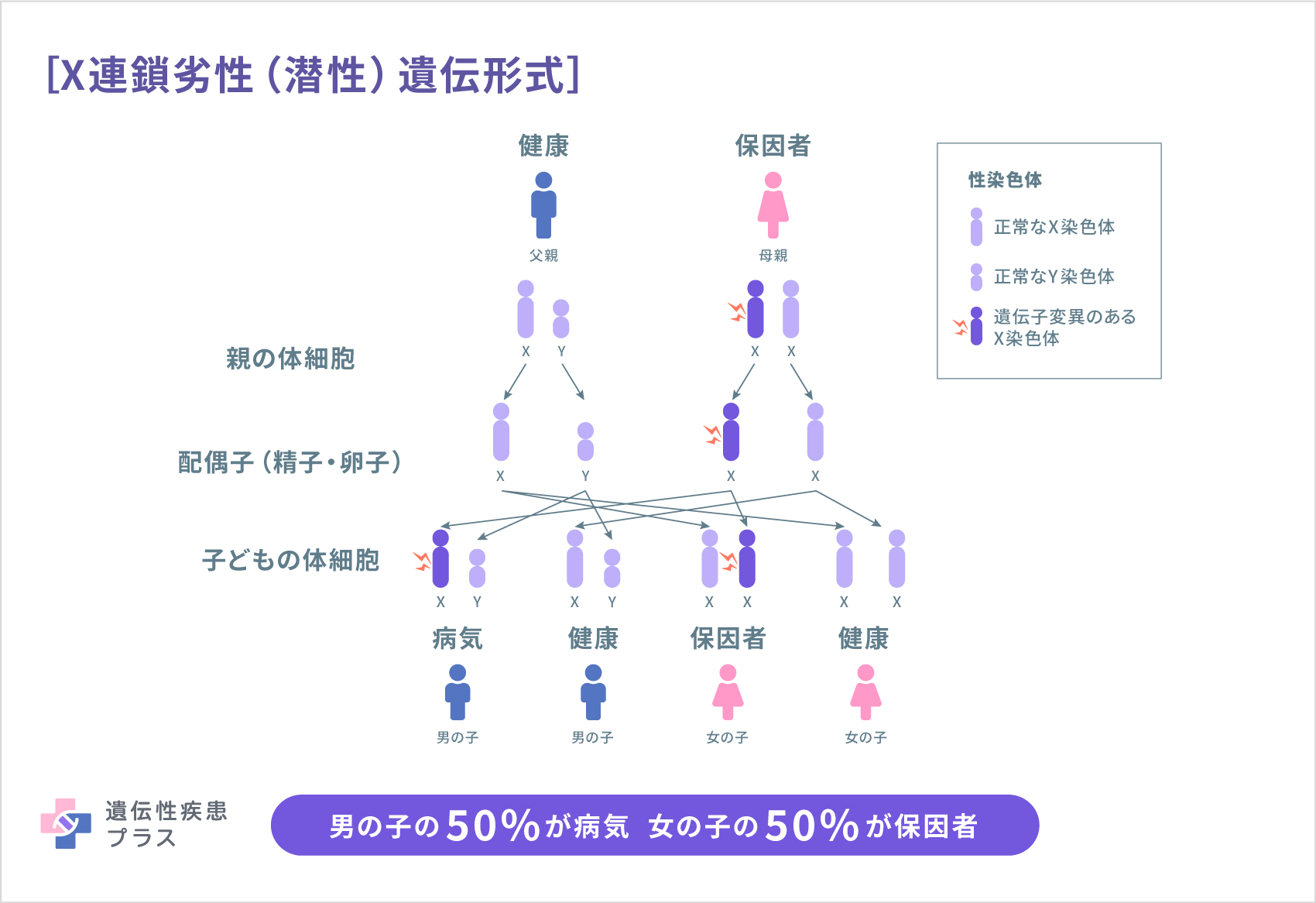
先天性角化不全症の遺伝形式は、原因遺伝子によって異なります。DKC1遺伝子が原因遺伝子となる場合、X連鎖劣性(潜性)遺伝形式で遺伝します。この遺伝形式では、男性(XY)である父親が変異を持つ場合、Y染色体は息子に、X染色体は娘に受け継がれ、息子は変異を受け継ぎませんが、娘には必ずその変異のあるX染色体が受け継がれます。女性(XX)である母親が変異を持つ場合は、変異の存在するX染色体が50%の確率で娘または息子に受け継がれます。男性はX染色体が1本のみであるため変異のあるX染色体を受け継ぐことでほとんどの場合発症します。一方、女性はX染色体を2本持っているため、通常はもう1本の正常な遺伝子が働くことで機能を補完し病気を発症しません。
また、原因遺伝子によって、常染色体劣性(潜性)遺伝形式や常染色体優性(顕性)遺伝形式などで遺伝します。


どのように診断されるの?
先天性角化不全症の診断基準によると、骨髄不全症(1系統以上の血球減少、骨髄低形成)を認め、さらに、皮膚、粘膜所見の大症状として、1)網状色素沈着、2)爪の萎縮、3)口腔粘膜白斑症、の3つのうち1つ以上、そして、その他の身体所見の小症状として、1)頭髪の喪失、白髪、2)歯芽の異常、3)肺病変、4)低身長、発育遅延、5)肝障害、6)食道狭窄、7)悪性腫瘍、8)小頭症、9)小脳失調、10)骨粗しょう症、の10個のうち2つ以上が認められることでこの病気であると診断されます。
また、遺伝学的検査で染色体テロメア長の制御に重要な遺伝子群DKC1、TERC、TERT、NHP2、NOP10、TINF2遺伝子などの変異の変異が同定されることでこの病気の診断が確定されます。
どのような治療が行われるの?
先天性角化不全症の根本的な治療法は確立されていないため、それぞれの症状に対する対症療法が中心となります。症状の重症度によって治療は異なり、軽症の場合には経過観察の場合もあります。
骨髄不全に対しては、再生不良貧血の重症度分類で中等症である場合には、ダナゾールなどの蛋白同化ホルモン(タンパク質同化ステロイド)という種類の薬が使われることがあります。蛋白同化ホルモンは腎臓に作用して赤血球産生を刺激するエリスロポエチンというホルモンを出させ、造血幹細胞にも直接作用して増殖を促すと考えられています。約半数程度の人において、一時的ではあるものの改善が見られるとされます。重症と判断される場合には、同種造血幹細胞移植が治療として選択される場合もあります。近年、同種造血幹細胞移植では、合併症を少なくするために骨髄非破壊的前処置が行われることがあります。
どこで検査や治療が受けられるの?
日本で先天性角化不全症の診療を行っていることを公開している、主な施設は以下です。
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
先天性角化不全症の患者会で、ホームページを公開しているところは、以下です。
参考サイト
- 難病情報センター
- 小児慢性特定疾病情報センター 先天性角化異常症
- MedlinePlus
- Genetic and Rare Diseases Information Center
- Online Mendelian Inheritance in Man(R) (OMIM(R))
- KEGG DISEASE:先天性角化異常症
- GeneReviews
- 厚生労働省科学研究費補助金 難治性疾患克服研究事業、先天性角化不全症の効果的診断法の確立と治療ガイドラインの作成に関する研究班「先天性角化不全症診療の参照ガイド」






