



どのような病気?
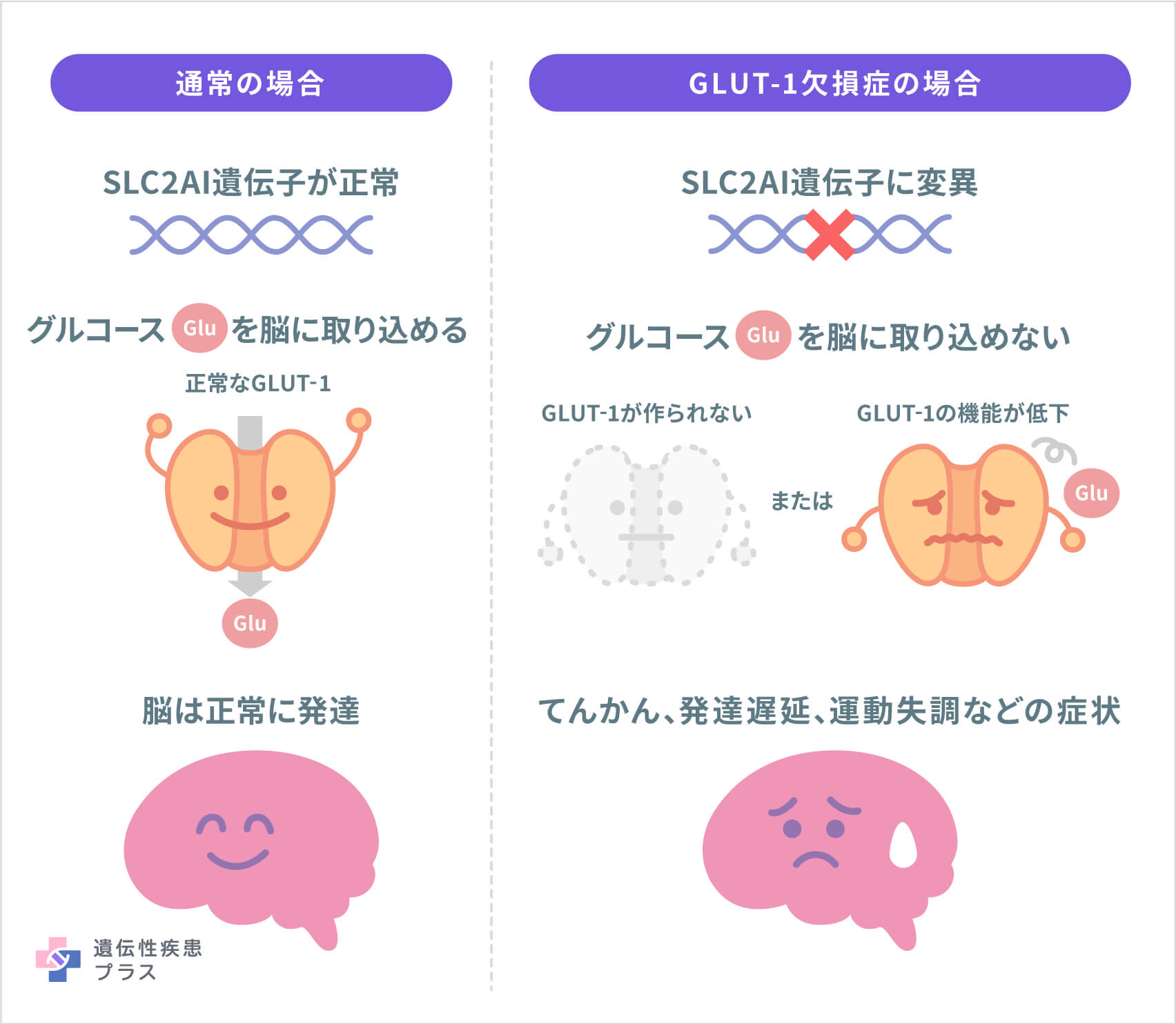
グルコーストランスポーター1(GLUT-1)欠損症(以下、GLUT-1欠損症)は、脳のエネルギー源であるグルコース(ブドウ糖)を脳内に取り込む仕組みに異常があることで生じる遺伝性疾患です。ヒトの体の細胞は、主にグルコースをエネルギー源としており、GLUT-1は細胞のグルコースの取り込みに関わるタンパク質です。この病気では、GLUT-1の産生に関わる遺伝子の異常によって、GLUT-1が作られない、または正しく機能できなくなり、脳にグルコースを必要な量取り込めなくなります。これにより、中枢神経系に起因するさまざまな症状が現れます。小児の脳は成人の3~4倍多くのグルコースが必要であるとされており、発達期の脳におけるグルコース不足は脳の機能や発達に大きな影響を及ぼします。遺伝子変異の種類により重症度が異なり、GLUT-1の機能が正常の75%以上残る遺伝子変異では軽微な症状のみが現れ、50~75%では軽症型となります。機能が50%程度に低下する遺伝子変異がGLUT-1欠損症の典型例とされます。
GLUT-1欠損症は、生まれてすぐには異常が見られません。最初の症状は乳児期に起こり、異常な目の動き、けいれん、無呼吸などが出現したり、消失したりします。その後、発達遅滞、痙性麻痺(けいせいまひ)、運動麻痺、頭痛、嘔吐、ジスキネジア、ジストニア、などのさまざまな症状がみられます。痙性麻痺とは脳などの中枢神経系が正常に働ないことによって手足が硬直して運動障害が起こることをいいます。ジスキネジアとは自分の意志とは関係なく、体の一部が勝手に不規則に動く現象(不随意運動)で、口がもぐもぐする、手が小刻みに常に動く、などがあります。ジストニアも自分の意志とは関係なく筋肉が収縮することによって姿勢の異常や硬直などが起こるものです。こうした症状は、空腹の時(特に早朝空腹時)や運動時、体温が上がった時、疲れた時に悪化し、食事、睡眠、安静などで改善します。また、症状にもよりますが、年齢とともに改善し、思春期以降は安定してくる傾向があります。症状のうち、てんかんは成長とともに軽減/消失する場合があります。また、ジスキネジア、痙性麻痺、運動失調などは思春期以降に出現/悪化する場合もありますが、命に別状はありません。
| GLUT-1欠損症で見られる症状 |
|---|
99~80%で見られる症状 運動失調、ジストニア、心電図異常、脳障害、発達遅延、小頭症、痙縮(筋緊張亢進状態)、てんかん、など |
79~30%で見られる症状 錯乱、チアノーゼ、言語発達遅延、構音障害、ジスキネジア、頭痛、麻痺、知的障害、無気力、筋硬直、不随意眼球運動発作、けいれん、など |
29~5%で見られる症状 中枢性無呼吸、間代性けいれん、睡眠障害、斜視、など |
割合は示されていないが見られる症状 代謝異常、反射亢進、発作性ジストニア、学習障害、など |
こうした症状のうち、見られる症状や重症度は人によってさまざまです。同じタイプの遺伝子変異を持っている人の間でも(同じ家系内に複数患者さんがいる場合も)、人によって異なりさまざまです。
また、GLUT-1欠損症では、髄液検査、血液検査、頭部の画像診断、脳波などに異常がみられます。髄液とは脳や脊髄の周囲を満たす液体でGLUT-1欠損症では髄液内のグルコース濃度の低下がみられます。血液検査では、低血糖がないにも関わらず髄液糖濃度の低下(40 mg/dL以下)や髄液糖/血糖比の低下(0.45以下)がみられます。頭部のCTやMRI検査により、大脳の萎縮がみられることがあります。また、脳波の異常がみられる場合もありますが、食事やグルコースの点滴で改善します。GLUT-1は赤血球の表面にも存在し、GLUT-1欠損症では赤血球のグルコースの取り込みも低下します。
GLUT-1欠損症は1991年にDe Vivo氏らによって初めて報告されたことから、De Vivo病(De Vivo disease)という別名もあります。最初の報告以来、世界で約500人の患者さんが報告されており、2011年に日本で行われた調査では50人以上が確認されました。国内にはまだ未診断の小児、成人例が多く存在すると考えられています。GLUT-1欠損症の正確な有病率は不明ですが、人種や性別によって差がないと考えられており、オーストラリアの報告では9万人に1人とされています。
GLUT-1欠損症は国の指定難病対象疾患(指定難病248)、および、小児慢性特定疾病の対象疾患です。
何の遺伝子が原因となるの?
多くの患者さんで1番染色体の1p34.2という位置にあるSLC2A1遺伝子に変異が見つかっています。SLC2A1遺伝子は、細胞のグルコースの取り込みに関わるタンパク質であるGLUT-1の設計図となるもので、この遺伝子の変異によってGLUT-1が作られなくなったり、その機能が低下したりします。
患者さんの多くは両親には遺伝子変異がなく、その人において新しく起こった変異(新生変異による孤発例)であることが、大規模な調査によりわかっています。一方で、家族例も複数報告されています。GLUT-1欠損症の人に子どもが生まれる場合、多くは常染色体優性(顕性)遺伝形式で遺伝します。この場合、両親のどちらかがGLUT-1欠損症の場合に子供が病気を受け継ぐ確率は50%です。

一方で、常染色体劣性(潜性)遺伝形式で遺伝している家系も少数報告されています。この場合、両親がともにSLC2A1遺伝子の片方に変異を持つ(保因者)場合、子どもは4分の1の確率でGLUT-1欠損症を発症します。また、2分の1の確率で保因者となり、4分の1の確率でこの遺伝子の変異を持たずに生まれます。

どのように診断されるの?
「新しい新生児代謝スクリーニング時代に適応した先天代謝異常症の診断基準作成と治療ガイドラインの作成および新たな薬剤開発に向けた調査研究班」が作成した、GLUT-1欠損症の診断基準は、以下です。
<GLUT-1欠損症が疑われる症状・検査所見>
空腹や運動によって増悪し、特に早朝空腹時に強く、食後に改善する下記の【症状】や【検査所見】があればGLUT-1欠損症が疑われます。
【症状】
乳児期早期発症のてんかん、発作性異常眼球運動、乳児期の筋緊張の低下、発達遅滞、痙性麻痺、ジストニア
【検査所見】
①血液検査
血糖値:正常
②髄液検査
髄液糖:40 mg/dL以下、髄液糖/血糖比0.45以下、髄液乳酸値は正常~低下
③脳波検査
背景脳波の徐波化(脳機能の低下を示す)がみられ、食後やグルコースの静脈注射によって脳波異常が改善する
④画像検査
頭部CTや頭部MRIで大脳萎縮、髄鞘化(脳の成長に伴って、脳の神経細胞同士が繋がって神経の伝達速度が向上する)の遅延などの所見がある
<GLUT-1欠損症の確定診断>
下記の1)~3)のいずれかに該当する場合にはGLUT-1欠損症と診断されます。
1)遺伝子検査でSLC2A1遺伝子の変異を認める
2)赤血球のグルコース取り込み試験でグルコースの取り込みが正常の60%以下
3)上記の1)及び2)に該当しない場合でも、上記【検査】の②髄液検査および③脳波検査の基準を満たし、ケトン食療法の導入から1か月以内に全ての症状が著明に改善
どのような治療が行われるの?
GLUT-1欠損症では「ケトン食療法」が行われます。これは、体内にケトン体を増やしやすくする食事療法です。ケトン食療法はてんかん発作やその他の症状の抑制に効果があり、知的能力、運動能力、意欲などを向上させることから、GLUT-1欠損症の診断がついたらなるべく早期に開始されます。乳児には、専用の調製粉乳が用いられます。
ヒトの体には、グルコースが不足したり利用できなかったりした場合に、グルコース以外のものをエネルギー源として利用する仕組みが備わっています。脂肪酸が分解される過程で生じる「ケトン体」は、グルコースの代わりのエネルギー源となり、また、細胞はGLUT-1の機能に関係なくケトン体を利用することができます。脂質はケトン体を作りやすく、糖質はケトン体を減らすように働くため、一般にケトン食は高脂肪食となります。ケトン食療法には、脂質やタンパク質、糖質の摂取量を計算して行う「古典的ケトン食」のほか、比較的管理が容易で負担が少ない「修正アトキンス食」などいくつかの種類があります。
ケトン食療法前は、GLUT-1の機能を抑えるような飲食物(カフェイン、アルコール)や薬剤(フェノバルビタール、抱水クロラール、テオフィリン)を摂取しないように注意します。また、ケトン食療法後は、逆に糖質制限が必要となります。過剰摂取で太ってしまったりしないように気をつけましょう。
そのほか、対症療法も行われます。例えば、てんかん発作に対しては抗てんかん薬の内服療法が行われます。また、障害された知的能力、身体能力を向上させ、社会適応能力を高めるためのリハビリテーションが行われます。
どこで検査や治療が受けられるの?
日本でGLUT-1欠損症の診療を行っていることを公開している、主な施設は以下です。
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
難病の患者さん・ご家族、支えるさまざまな立場の方々とのネットワークづくりを行っている団体は、以下です。
米国のGLUT-1欠損症の患者支援団体(財団)で、ホームページを公開しているところは、以下です。
参考サイト
- 難病情報センター
- 小児慢性特定疾病情報センター グルコーストランスポーター1(GLUT1)欠損症
- MedlinePlus
- Genetic and Rare Diseases Information Center






