



どのような病気?
遺伝性鉄芽球性貧血は、遺伝子の変異が原因で鉄がうまく利用できなくなり、貧血症状と鉄の蓄積による心臓や肝臓の障害などの症状が引き起こされる遺伝性疾患です。
鉄芽球性貧血とは、骨髄(血液細胞を作る組織)において、赤血球になる前の段階の「赤芽球」と呼ばれる細胞の核の周囲に鉄が環状に沈着する(環状鉄芽球)ことを特徴とする貧血です。鉄芽球性貧血は、遺伝子変異が原因の遺伝性のものと、骨髄異形成症候群と呼ばれる疾患や薬物などを原因とする後天的なものに大きく分けられます。この記事では、遺伝子変異が原因の「遺伝性鉄芽球性貧血」について説明します。
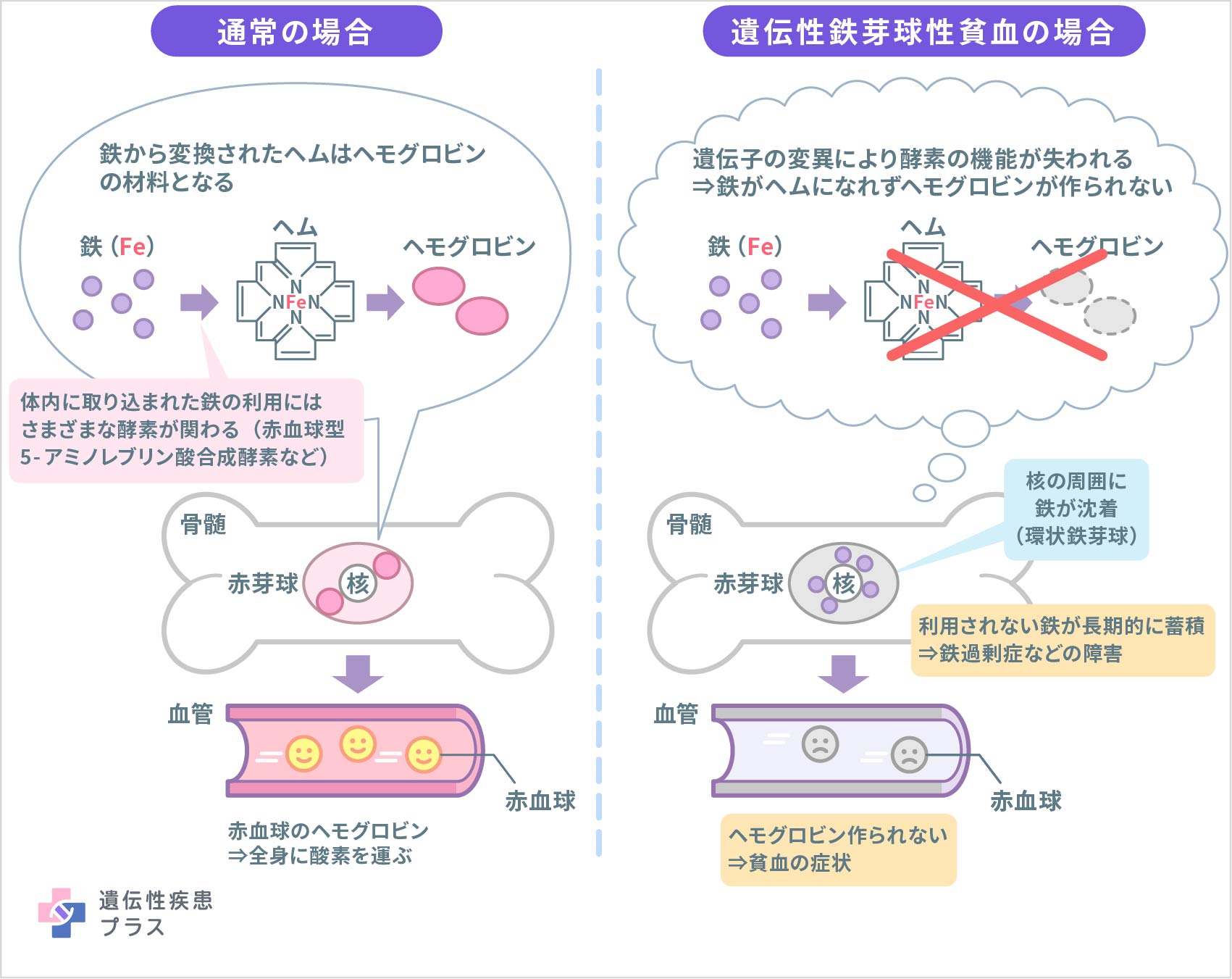
貧血には、赤血球に含まれる「ヘモグロビン」と呼ばれるタンパク質が関連します。赤血球は、ヘモグロビンに酸素を結合することで、全身に酸素を送りますが、そのヘモグロビンを作るためには鉄が必要です。この病気の貧血の原因は病型によって異なりますが、原因の一つとして赤血球ができる際に細胞の中にある鉄をうまく利用できずヘモグロビンが作られないことによる貧血があります。
この病気の貧血は、病型によって重症度や発症年齢が異なり、重症度は軽症~重症まで幅広く認められます。主に、顔色の悪さ、息切れ、動悸、めまい、頭痛、疲れやすさなど、貧血による症状が引き起こされます。発症時期はさまざまですが、多くの場合若年期から貧血症状が見られることが多いとされます。
また、うまく利用されない鉄が体に長期的に蓄積することや輸血などによって、鉄過剰症(ヘモクロマトーシス)が合併し、肝臓と脾臓の肥大、肝障害、糖尿病、心不全などが認められることがあります。ヘモクロマトーシスは、病型と輸血の量などによっても重症度が異なります。
遺伝性鉄芽球性貧血のほとんどはX連鎖性遺伝形式をとる鉄芽球性貧血1(XLSA)という病型で、X連鎖性鉄芽球性貧血(X-linked sideroblastic anemia)とも呼ばれます。XLSAは、ほとんどの場合男性に認められますが、まれに女性でも見られることがあります。XLSAで貧血や倦怠感以外に高頻度に見られる症状としては、筋力低下や蒼白などがあり、しばしば見られる症状には、呼吸困難、皮膚の色素沈着過剰、脾臓の肥大(脾腫)などがあります。それ以外の病型は非常にまれですが、病型によって貧血の重症度や発症年齢はさまざまです。さらに症候性と呼ばれる、貧血だけでなく神経症状、筋症状などの全身症状が見られる病型もあります(下表、遺伝性鉄芽球性貧血の主な病型と特徴を参照)。
遺伝性鉄芽球性貧血の主な病型と特徴
非症候性(鉄芽球性貧血以外の症状がほとんど見られないとされる病型)
病型 | 発症年齢 | 貧血の程度 | 合併症など |
|---|---|---|---|
鉄芽球性貧血1(XLSA) | さまざま | 軽~重症 | 鉄過剰症 |
鉄芽球性貧血2(SA/SLC25A38) | 小児 | 重症 | 鉄過剰症 |
鉄芽球性貧血3(SA/GLRX5) | 成人 | 軽~重症 | 鉄過剰症 |
鉄芽球性貧血4 (SA/HSPA9) | さまざま | 軽~中等症 | 不明 |
症候性(鉄芽球性貧血以外のさまざまな症状が合併する病型)
病型 | 発症年齢 | 貧血の程度 | 合併症など |
|---|---|---|---|
小脳失調を伴うX連鎖性鉄芽球性貧血(XLSA/A) | 幼児期 | 軽~中等症 | 小脳性運動失調など |
ミトコンドリアDNA異常に伴うPearson-marrow-pancreas症候群(PMPS) | 乳児期まで | 重症 | 代謝性アシドーシス、外分泌機能低下、腎不全 |
チアミン反応性巨赤芽球性貧血(TRMA) | さまざま | 重症 | 巨赤芽球性貧血、糖尿病、神経性難聴 |
筋症、乳酸アシドーシスおよび鉄芽球性貧血(MLASA1/PUS1) | 小児 | 軽~重症 | 乳酸アシドーシス、筋症、心筋症 |
筋症、乳酸アシドーシスおよび鉄芽球性貧血(MLASA2/YARS2) | 小児 | 軽~重症 | 乳酸アシドーシス、筋症、心筋症 |
筋症、乳酸アシドーシスを伴う鉄芽球性貧血(MLASA3/ATP6) | 小児 | 中等度~重症 | 代謝性アシドーシス、筋症、神経系異常 |
水腫、乳酸アシドーシスおよび鉄芽球性貧血(HLASA/LARS2) | 小児 | 重症 | 乳酸アシドーシス、けいれん、心臓機能不全、水腫、発達遅滞など |
遺伝性鉄芽球性貧血とともにB細胞の欠損、周期性発熱、発育障害を示す症候群(SFID/TRNT1) | 小児 | 重症 | 鉄過剰症、難聴、発熱、心筋症、発達障害 |
NDUFB11の変異に伴う先天性鉄芽球性貧血(SA/NDUFB1) | さまざま | 軽~中等症 | 乳酸アシドーシス |
遺伝性鉄芽球性貧血は、まれな疾患のため発症頻度や患者数ははっきりわかっていません。
厚生労働省研究班「遺伝性鉄芽球性貧血の診断分類と治療法の確立班」による国内の遺伝性鉄芽球性貧血の実態調査では、変異遺伝子が確定した患者さんの大多数はXLSAであり、ミトコンドリアDNA異常に伴うPearson-marrow-pancreas症候群(PMPS)や、鉄芽球性貧血2(SA/SLC25A38)も見つかっています。
遺伝性鉄芽球性貧血は、国の指定難病対象疾患(指定難病286)、および後天的な鉄芽球性貧血を含めた鉄芽球性貧血として小児慢性特定疾病の対象疾患となっています。
何の遺伝子が原因となるの?
遺伝性鉄芽球性貧血では、これまでに複数の原因遺伝子が報告され、原因遺伝子ごとに病型が分かれています(下表参照)。
遺伝性鉄芽球性貧血の原因遺伝子
病型 | 原因遺伝子 | 染色体などの領域 | 作られるタンパク質など | 遺伝形式 |
|---|---|---|---|---|
鉄芽球性貧血1(XLSA) | ALAS2 | Xp11.21 | 赤血球型5(δデルタ)-アミノレブリン酸合成酵素 | X連鎖劣性(潜性) |
鉄芽球性貧血2(SA/SLC25A38) | SLC25A38 | 3p22.1 | 溶質キャリアファミリー25メンバー38 | 常染色体劣性(潜性) |
鉄芽球性貧血3(SA/GLRX5) | GLRX5 | 14q32.13 | グルタレドキシン5 | 常染色体劣性(潜性) |
鉄芽球性貧血4(SA/HSPA9) | HSPA9 | 5q31.2 | ミトコンドリア熱ショックタンパク質A9 | 常染色体劣性(潜性)または常染色体優性(顕性)、偽優性(顕性)遺伝の報告もある |
小脳失調を伴うX連鎖性鉄芽球性貧血(XLSA/A) | ABCB7 | Xp13.3 | ATP結合カセットサブファミリーBメンバー7 | X連鎖劣性(潜性) |
ミトコンドリアDNA異常に伴うPearson-marrow-pancreas症候群(PMPS) | mtDNA(1,000~1万塩基の欠失) | MtDNAの欠失 | - | 母系遺伝(孤発例も多い) |
チアミン反応性巨赤芽球性貧血(TRMA) | SLC19A2 | 1q24.2 | 溶質キャリアファミリー19メンバーA2 | 常染色体劣性(潜性) |
筋症、乳酸アシドーシスおよび鉄芽球性貧血(MLASA1/PUS1) | PUS1 | 12q24.33 | シュードウリジン合成酵素1 | 常染色体劣性(潜性) |
筋症、乳酸アシドーシスおよび鉄芽球性貧血(MLASA2/YARS2) | YARS2 | 12p11.21 | チロシルtRNA合成酵素2 | 常染色体劣性(潜性) |
筋症、乳酸アシドーシスを伴う鉄芽球性貧血(MLASA3/ATP6) | MT-ATP6 | MtDNA(8527~9207) | ATPase 6(ATP合成酵素の一部) | 母系遺伝(孤発例も多い) |
水腫、乳酸アシドーシスおよび鉄芽球性貧血(HLASA/LARS2) | LARS2 | 3p21.31 | ロイシルtRNA合成酵素 | 常染色体劣性(潜性) |
遺伝性鉄芽球性貧血とともにB細胞の欠損、周期性発熱、発育障害を示す症候群(SFID/TRNT1) | TRNT1 | 3p26.2 | tRNAヌクレオチジルトランスフェラーゼ | 常染色体劣性(潜性)遺伝形式 |
NDUFB11の変異に伴う先天性鉄芽球性貧血(SA/NDUFB1) | NDUFB11 | Xp11.3 | ミトコンドリアNADH-ユビキノン酸化還元酵素 | X連鎖性遺伝形式 |
遺伝性鉄芽球性貧血で患者さんが最も多い病型は、X染色体のXp11.21と呼ばれる領域に存在するALAS2遺伝子を原因とする鉄芽球性貧血1(XLSA)です。ALAS2遺伝子は、ヘムと呼ばれる分子の生成に重要な赤血球型5-アミノレブリン酸合成酵素と呼ばれる酵素タンパク質の設計図となります。
ヘモグロビン生成のためには、取り込まれた鉄がミトコンドリアと呼ばれる細胞小器官においてヘムと呼ばれる分子に変換されることが必要になりますが、遺伝子の変異によって、赤血球型5-アミノレブリン酸合成酵素の機能が損なわれると、正常なヘムの生成ができなくなります。そしてヘモグロビンが不足して貧血症状が引き起こされます。また、赤芽球に送られてきた鉄のほとんどが本来はヘムになるところで、ヘムが作られないために、細胞の中に過剰な鉄がどんどん蓄積します。さらに、貧血を感知した体はヘモグロビンを作らせるために食事からより多くの鉄を吸収しようとします。こうして蓄積された過剰な鉄は、臓器に損傷を与え、XLSAの症状が引き起こされると考えられています。
その他の病型の遺伝性鉄芽球性貧血においても、ALAS2遺伝子と同様にヘムと呼ばれる分子の変換に関わるもののほか、赤血球におけるヘムや鉄の代謝に関わる遺伝子、ミトコンドリアのタンパク質合成に関わる遺伝子などが報告されており、多くはミトコンドリアにおける鉄の利用がうまくいかないことが、環状鉄芽球を生じさせる原因と考えられています。しかし、それぞれの原因遺伝子の変異がなぜ貧血の原因となるのかはっきりわかっていない病型もあります。
上記以外にも、ヘムや鉄の代謝に関わるSTEP3、FECH、HSC20遺伝子などがこの病気の原因遺伝子として報告されているほか、まだ原因遺伝子が不明の遺伝性鉄芽球性貧血もあります。まれな例として、海外では上記の変異に加えてHFE遺伝子に変異がある場合にヘモクロマトーシスが重症化するという報告があります。
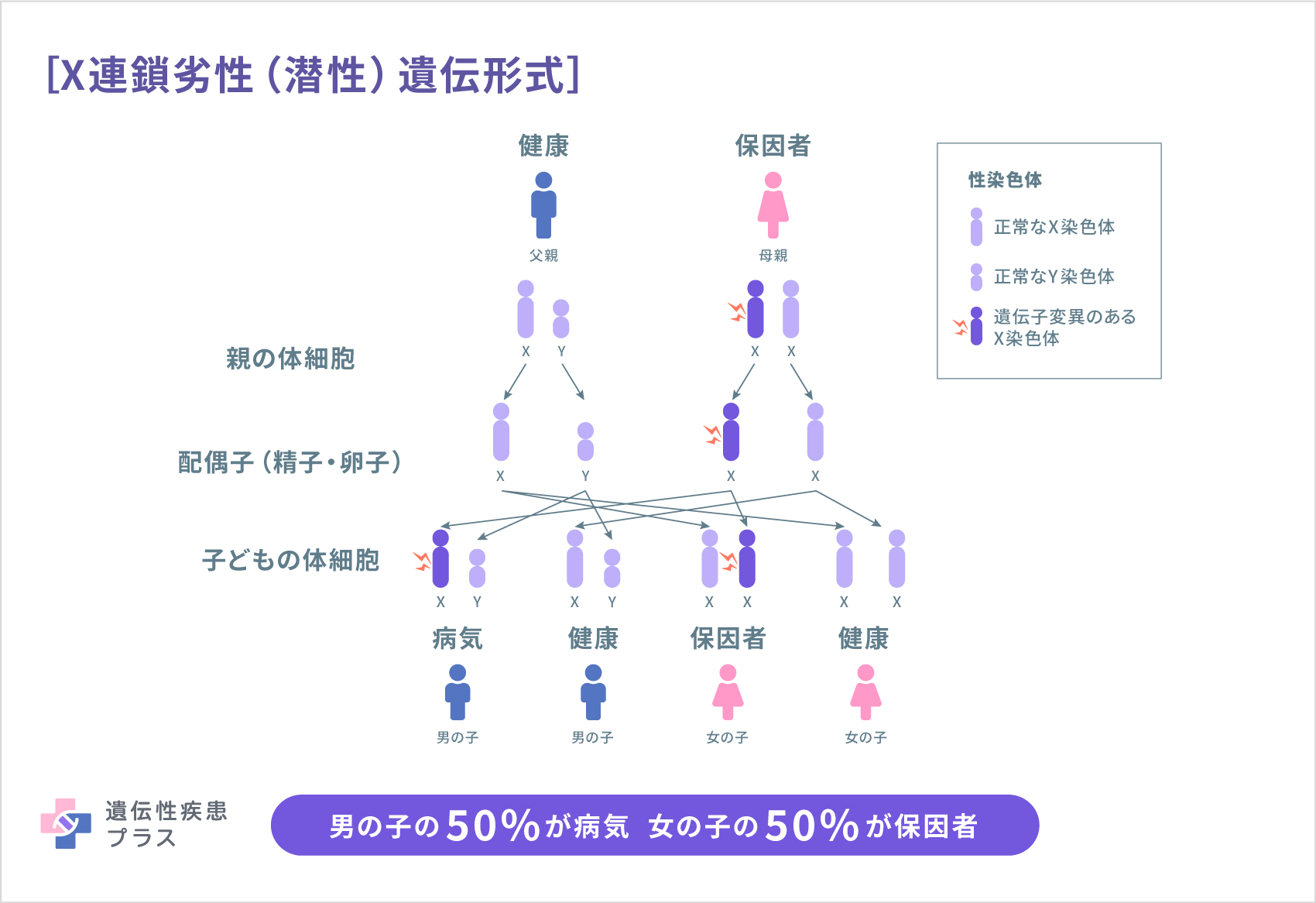
遺伝性鉄芽球性貧血の遺伝形式は、原因遺伝子によって異なります。ALAS2遺伝子が原因遺伝子である鉄芽球性貧血1(XLSA)では通常、X連鎖劣性(潜性)遺伝形式で遺伝します。この遺伝形式では、病気の遺伝する確率が性別によって異なります。男性(XY)である父親が変異を持つ場合、Y染色体は息子に、X染色体は娘に受け継がれるため、息子は変異を受け継ぎませんが、娘には必ずその変異のあるX染色体が受け継がれることになります。女性(XX)である母親が変異を持つ場合は、変異の存在するX染色体が50%の確率で娘または息子に受け継がれることになります。男性はX染色体が1本のみですが、女性はX染色体を2本持っているため、通常はもう1本の正常な遺伝子が働くことで機能を補完します。この遺伝形式ではほとんどの場合、男性だけが発症し、女性は保因者となり病気を発症しませんが、XLSAではまれに女性にも発症することが報告されています。
また、原因遺伝子によっては、常染色体劣性(潜性)遺伝形式や常染色体優性(顕性)遺伝形式で遺伝します。


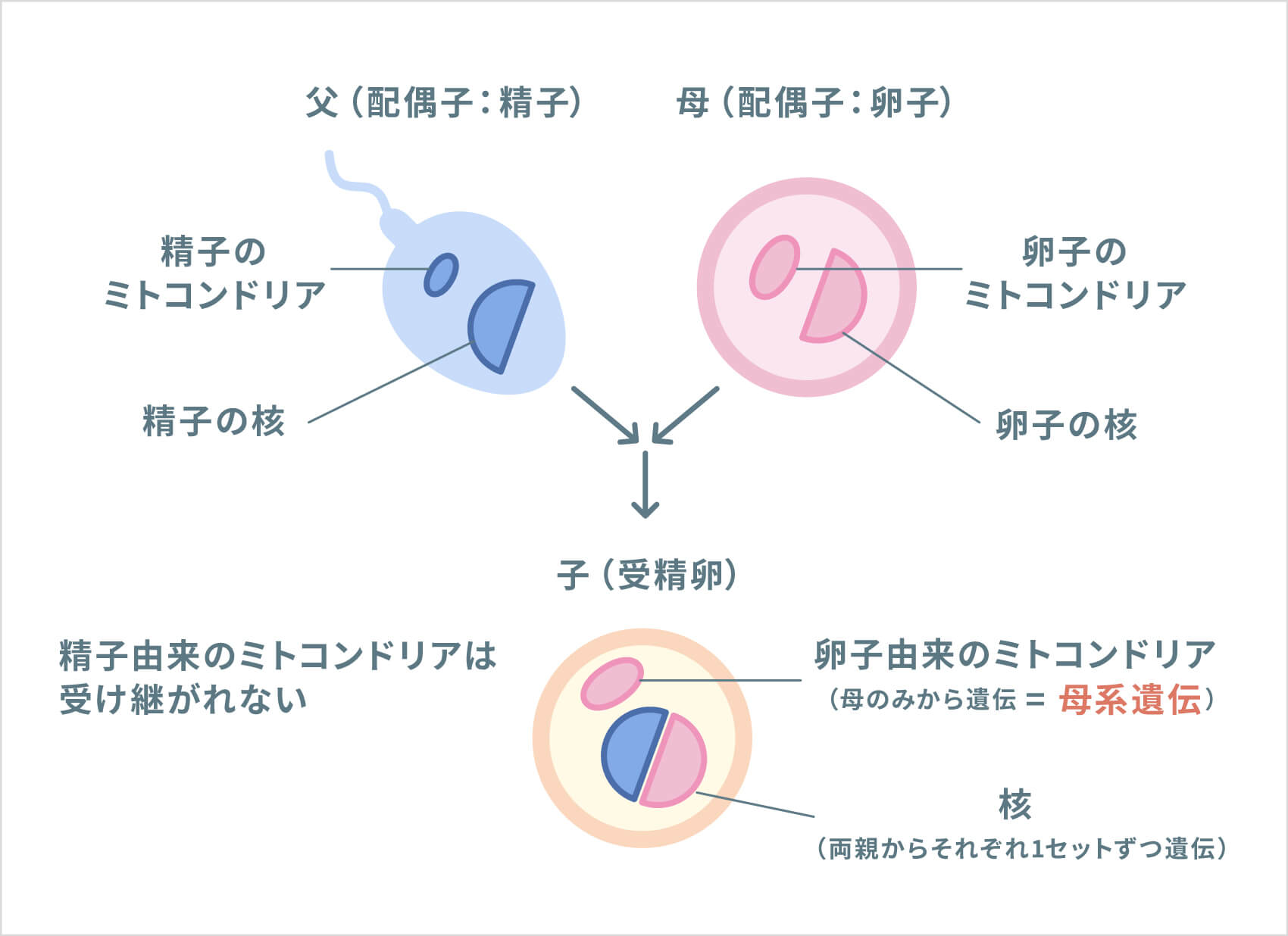
一方、ミトコンドリア遺伝子の変異が原因となる場合は、「母系遺伝」と呼ばれる遺伝形式となります。受精卵のミトコンドリアは、全て母親の卵子に含まれていたミトコンドリアとなり、精子のミトコンドリアは、子どもの細胞に受け継がれないため、病気の父親から子供へは遺伝しません。母親のミトコンドリアDNAに変異があった場合、子どもに病気が受け継がれる可能性があります。
どのように診断されるの?
難病情報センターによれば、この病気の診断基準として、検査の結果、1)貧血(男性ヘモグロビン<13g/dL、女性ヘモグロビン<12g/dL)、2)骨髄にて環状鉄芽球の出現(15%以上)、3)血清鉄の上昇、UIBCの低下、血清フェリチンの上昇、の1)~3)を認めること、さらに遺伝学的検査においてALAS2、SLC25A38、PUS1、ABCB7、GLRX5、SLC19A2などの遺伝子、ミトコンドリアDNAにおいて変異が認められた場合にはこの病気の診断が確定されます。
また、遺伝学的検査の結果に関わらず、小児期に発症し、かつ骨髄異形成症候群、薬剤性、アルコール性などの二次性鉄芽球性貧血、他の先天性貧血(サラセミアなど)の疾患ではないことが確認(鑑別診断)され、さらに家族歴(血縁関係のある人に同じ病気がある)を持つ場合にはこの病気の可能性ありと診断されます。
どのような治療が行われるの?
遺伝性鉄芽球性貧血のうち、ALAS2遺伝子を原因とする鉄芽球性貧血1(XLSA)については、赤血球型5-アミノレブリン酸合成酵素の働きを助けるビタミンB6を投与する治療が、半数の患者さんに有効で貧血の改善が認められることがわかっています。また、チアミン反応性巨赤芽球性貧血(TRMA)では、ビタミンB1投与治療が有効な場合があります。
それ以外の病型については治療法が確立されていません。貧血が長期に渡る場合には、過剰な鉄を除去する除鉄療法や、重症な場合には造血幹細胞移植が行われることもあります。
どこで検査や治療が受けられるの?
日本で遺伝性鉄芽球性貧血の診療を行っていることを公開している、主な施設は以下です。
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
難病の患者さん・ご家族、支えるさまざまな立場の方々とのネットワークづくりを行っている団体は、以下です。
参考サイト
- 難病情報センター
- 小児慢性特定疾病情報センター 鉄芽球性貧血
- MedlinePlus
- Genetic and Rare Diseases Information Center
- Online Mendelian Inheritance in Man(R) (OMIM(R))
- 遺伝性鉄芽球性貧血診療の参照ガイド 令和 1 年改訂版






