



どのような病気?
低ホスファターゼ症は、「組織非特異的アルカリホスファターゼ(TNSALP)」という酵素が体内で正常にはたらかないことにより、骨や歯が弱くなるのが特徴的な、遺伝性疾患です。「組織非特異的」というのは、いろいろな組織に幅広く存在しているという意味ですが、TNSALPは、特に骨、歯、肝臓、腎臓、血液で活性がみられます。
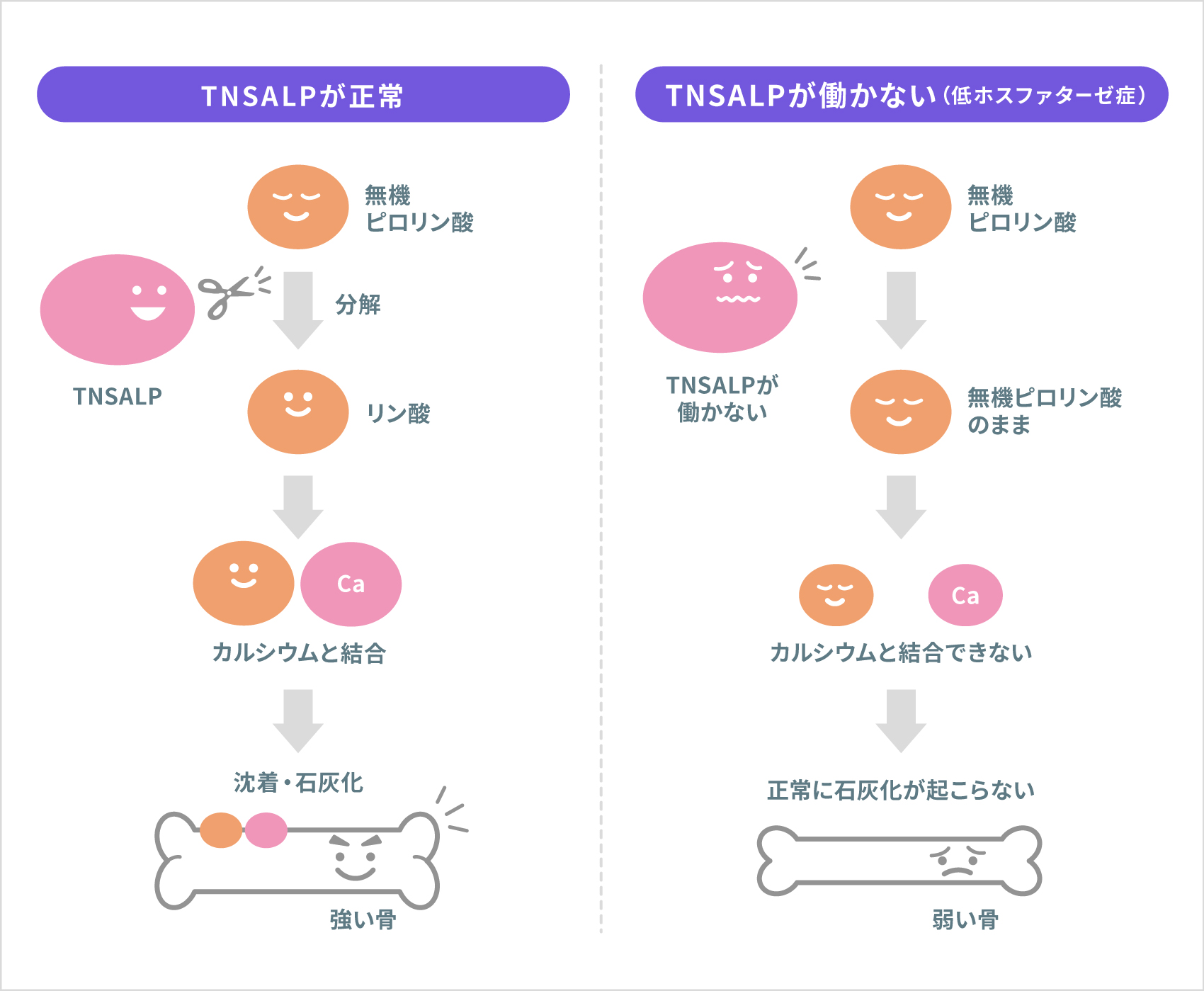
「ホスファターゼ」とは、物質から「リン酸」を含む部分を取り出すはたらきをする酵素の総称です。TNSALPは、骨や歯が作られる過程で、「無機ピロリン酸」を分解して「リン酸」を取り出します。このリン酸とカルシウムが結合して骨や歯に沈着すると、強くて硬い骨や、硬いものを噛み砕ける歯など、正常な状態に発達します(これを石灰化といいます)。低ホスファターゼ症の人は、TNSALPがうまくはたらかない、または完全にはたらかなくなっているため、骨や歯を硬くする「石灰化」が正常に起こらず、病気を発症します。無機ピロリン酸が分解されずに蓄積することが、石灰化を妨げているとの研究報告もあります。
低ホスファターゼ症は、お母さんのおなかの中にいるうちから発症する人もいれば、成人になってから発症する人もおり、発症年齢はさまざまです。乳歯が通常より早く(5歳までに)抜け始める人も多く、この場合、抜けた乳歯には、歯根が残っているという特徴がみられます。治療をしなかった場合、生存が難しいほど重症の人から、ほとんど症状のない人まで、重症度もさまざまです。こうした発症時期や、重症度により、6つの病型に分類されています。
低ホスファターゼ症の6つの病型と特徴
| 病型 | 遺伝形式 | 特徴 |
|---|---|---|
| 周産期重症型 | 常染色体劣性(潜性)遺伝 | 妊娠22週から出生後7日未満までの期間(周産期)に発症が確認される、最も重症な病型。出生時に手足が短い、頭囲が相対的に大きい、胸郭が狭くなり呼吸障害を起こす、などがみられる。X線検査をすると、全身の骨が低石灰化を起こしている、長管骨(手足の細長い形の骨)が変形している、などがみられる。 |
| 周産期軽症型 | 常染色体劣性(潜性)遺伝または常染色体優性(顕性)遺伝 | 骨の変形などから胎児期に低ホスファターゼ症と診断された人のうち、骨の石灰化が良好におきており、経過が良好な病型。日本人では、ALPL遺伝子変異のうち「F310L」という変異との関連が指摘されている。 |
| 乳児型 | 多くは常染色体劣性(潜性)遺伝 | 生後6か月までに発症する。乳児期に死亡する場合もある経過が不良な病型。頭蓋骨の隙間が早くくっついてしまう「頭蓋骨縫合早期癒合症」、低石灰化、幼児くる病、高カルシウム尿症、哺乳不良、体重増加不良などがみられる。 |
| 小児型 | 常染色体劣性(潜性)遺伝または常染色体優性(顕性)遺伝 | 小児期に発症し、重症度はさまざま。乳歯が早期(4歳くらいまで)に抜け落ちるのが特徴。低身長、骨格異常、骨痛、病的な骨折などがみられる。 |
| 成人型 | 常染色体劣性(潜性)遺伝または常染色体優性(顕性)遺伝 | 成人期(おもに中年期)になってから発症。病的に骨折しやすい、骨や関節が痛む、永久歯が抜けやすい、などの特徴がある。 |
| 歯限局型 | 常染色体劣性(潜性)遺伝または常染色体優性(顕性)遺伝 | 歯に限局して症状がみられる。乳歯が早期に抜け落ちる、歯の土台となる骨(歯槽骨)が喪失している、などがみられる。 |
その他、骨の石灰化が妨げられている結果カルシウムが血中に多く存在し、高カルシウム血症/尿症や、腎結石がみられる人や、TNSALPの、ビタミンB6を正しく使えるようにするためのはたらきが妨げられるために、脳がビタミンB6欠乏状態となり、けいれんを起こす人などもいます。
この病気の人が生まれる頻度は、15万人に1人程度とされ、重症で命を落とす人もいることから、患者さんの数は国内に100~200人程度と推定されてきました。しかし、有効な薬が開発され2015年に承認されたことにより、これまで命を落としていた人も救えるようになりました。そのため、患者さんの数は、少しずつ増えていると考えられています。低ホスファターゼ症は、小児慢性特定疾病、および、指定難病対象疾病です(指定難病172)。
何の遺伝子が原因となるの?
低ホスファターゼ症の人は、「ALPL遺伝子」に異常があることがわかっています。ALPL遺伝子は、「組織非特異的アルカリホスファターゼ(TNSALP)」の設計図となる遺伝子です。この遺伝子は、第1染色体の短腕の、1p36.12という位置に存在しており、男女差などは病気のなりやすさに影響しません。
低ホスファターゼ症の人では、ALPL遺伝子のどの部分が変異しているのかというと、これは人によってさまざまで、約300箇所も確認されています。これらの変異のほとんどは、TNSALPを構成するアミノ酸の1つを変化させるような変異です。その他、TNSALPの構造を大きく変化させるような変異もあります。変異により、TNSALPのはたらきがどの程度弱まるかが、重症度に関与していると考えられています。つまり、TNSALPが全くはたらかないような変異は重症になり、はたらきを弱める程度の変異は、軽症になると考えられています。しかし、これらの多くの変異と重症度の関連性は、まだ完全には解明されていません。
この病気は親から子へ遺伝しますが、遺伝のしかた(遺伝形式)は、「常染色体劣性(潜性)遺伝」または「常染色体優性(顕性)遺伝」です。それぞれの病型における遺伝形式は、前述の表のとおりです。
常染色体劣性(潜性)遺伝の場合、たまたま両親ともに、1人あたり2つセットでもつALPL遺伝子の片方(1つ)が変異しているけれども無症状、という人で、その子どもが親から1つずつ変異した遺伝子を受け継ぎ、2つとも変異した状態になった場合に発症します。常染色体優性(顕性)遺伝の場合は、2つもつALPL遺伝子の1つに変異があれば発症します。


どのように診断されるの?
低ホスファターゼ症には、医師が低ホスファターゼ症と診断するための「診断基準」があります。したがって、病院へ行き、必要な問診や検査を受けた後、主治医の先生がそれらの結果を診断基準に照らし合わせ、結果的に低ホスファターゼ症かそうでないかの診断をすることになります。具体的には、X線検査で骨の石灰化障害がみられた、または乳歯が4歳未満で抜け落ちた、という症状(主症状)の片方あるいは両方がみられ、血液検査でアルカリホスファターゼ(ALP)の値が低かった場合(正常値は年齢によって異なる)には、低ホスファターゼ症の疑いとなり、ALPL遺伝子の遺伝子検査を受けて変異があれば確定診断となります。
その他、参考症状として、ビタミンB6依存性けいれん、手足の短縮や変形が挙げられ、参考検査所見として、尿中ホスフォエタノールアミンの上昇、血清ピロリン酸値の上昇、乳児における高カルシウム血症が挙げられます。また、参考所見として、家族歴や、両親の血清ALP値の低下も挙げられます。
重症度分類は、食事・栄養、呼吸のそれぞれの評価スケールに基づいてなされます。
どのような治療が行われるの?
以前は、根本的な治療法がなく、症状を緩和する「対症療法」しかありませんでした。対症療法では、呼吸が十分にできない人が人工呼吸器を使ったり、骨が痛い人は痛み止めを使ったりします。また、重症型でけいれんを起こす人には、ビタミンB6が投与されたり、乳児型でしばしばみられる高カルシウム血症には、低カルシウムミルクが使用されたりします。
2015年に、ALPを注射で体内に補充する「酵素補充療法」が日本でも承認され、根本的な治療法として使えるようになりました。特に、重症の患者さんでは、酵素補充療法が使用されます。酵素補充療法が行われるようになったことで、救われる命が増えることが期待されています。
また、骨髄の幹細胞を移植する、細胞療法の臨床試験が、国内で複数進められています。幹細胞は、さまざまな組織に分化可能な細胞で、骨髄などヒトの体に多数存在しています。これを移植することでALPを作る細胞を定着させて、骨の形成を正常にしていくことが狙いの治療法です。酵素補充療法のように定期的に注射を行う必要が無い、根本的な治療法として注目されています。
遺伝子治療については、まだ動物実験で検討が行われている段階です。
ALPは、一般的に成長とともに必要性が減っていきます。そのため、軽症の人では自然に症状が改善する場合もあります。
日常生活では、症状がほとんどないという人も、骨折や歯周病などを起こしやすい可能性があるので、定期的に診察を受けて、骨の痛みや歯の健康などに注意します。
どこで検査や治療が受けられるの?
日本で低ホスファターゼ症の診療を行っていることを公開している、主な施設は以下です。
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
低ホスファターゼ症の患者会で、ホームページを公開しているところは、以下です。
HPP HOPE 低ホスファターゼ症コミュニティの取材記事は、以下です。
参考サイト
・参考文献:医学書院 医学大辞典 第2版







