



どのような病気?
メチルマロン酸血症はメチルマロン酸をはじめとする酸性物質が体内に蓄積することでさまざまな症状を来す疾患です。メチルマロン酸は体内でエネルギー産生などに関与する物質です。メチルマロン酸に補酵素A(CoA)が結合したメチルマロニルCoAはメチルマロニルCoAムターゼ(MCM)という酵素で代謝されますが、MCMの活性が低下することで、メチルマロン酸が利用されずに組織に蓄積し、メチルマロン酸血症が発症します。症状の重症度はMCMの活性の程度に影響されます。
MCMの活性が低下する要因としては、MCM欠損症(正しく働くMCMが作られない、もしくはMCMそのものが作られない)とMCMの補酵素として作用するコバラミン(ビタミンB12の1つの型)の合成障害があります。
コバラミンの合成障害にはcblA~Gという7つのタイプがあります。cblAおよびcblBのタイプはMCMの補酵素として作用するコバラミンの1つであるアデノシルコバラミンの合成障害によるものです。cblC、cblE、cblF、cblGの各タイプはアデノシルコバラミンの合成も障害されますが、ホモシステインという物質からメチオニンを合成する際に必要なメチルコバラミンの合成も障害されるもので、メチルマロン酸の増加に加えて、ホモシステインも増加します。ホモシステインが増加するとホモシステインが2つ結合したホモシスチンが産生され、ホモシスチン尿症が見られます。最後のcblDタイプはメチルマロン酸のみが増加する病型(メチルマロン酸血症単独型)、ホモシスチン尿症のみが見られる病型(ホモシスチン尿病単独型)、その両方が見られる病型(混合型)の3つがあります。以上のようにメチルマロン酸血症にはMCM欠損症とコバラミンの合成障害による7つのタイプを併せて8つのタイプがあります。この記事では主に、メチルマロン酸血症について、MCM欠損症、コバラミン合成障害タイプcblA、cblBおよびcblDのうちホモシステイン増加が見られない病型の4つを対象として記載しています。
メチルマロン酸血症では、急性代謝不全、中枢神経系症状、腎機能障害などが見られます。急性代謝不全としては、新生児期から乳児期におけるケトアシドーシスや高アンモニア血症などが発現することがあります。ケトアシドーシスとは脂肪の代謝産物であるケトンが体内に過剰に蓄積することで血液のpHが低下し、血液が酸性に傾く状態です。ケトアシドーシスや高アンモニア血症が生じると哺乳不良・嘔吐・呼吸障害・筋緊張低下などが発現し、急性脳症に進展することがあります。回復した後も同様の急性増悪を繰り返しやすく、特に感染症への罹患などが急性増悪の契機となります。コントロールが困難な場合には摂食不良によって身体発育が遅延する場合があります。また、急性代謝不全の後遺症や病気の進行などによって精神運動発達遅滞を生じることがあります。そのほか、腎機能障害、心筋症、膵炎などが見られることがあります。
メチルマロン酸血症の有病率は海外では5万人から10万人に1人程度とされています。日本の調査では11万人に1人程度とされ、国内の患者さんは約300人とされています。
何の遺伝子が原因となるの?
メチルマロン酸血症がMCM欠損症による場合にはその原因遺伝子はMUT、コバラミン合成障害の場合にはその原因遺伝子はcblAではMMAA、cblBではMMAB、cblDではMMADHCになります。メチルマロン酸血症の患者さんのおよそ60%はMUT遺伝子の変異によります。MUTの変異によって、機能するMCMが全く産生されないタイプはメチルマロン酸血症の最重症型であり、予後不良です。MUTの変異があっても、ある程度の機能するMCMが産生される場合には重症度は低下します。
また、海外を中心にホモシスチン尿症を伴わない単独型のメチルマロン酸血症の他の原因としてMCEE遺伝子の変異も報告されています。この遺伝子は、メチルマロニル CoA エピメラーゼと呼ばれる酵素の設計図となり、メチルマロニル CoA エピメラーゼ欠損症とも呼ばれます。
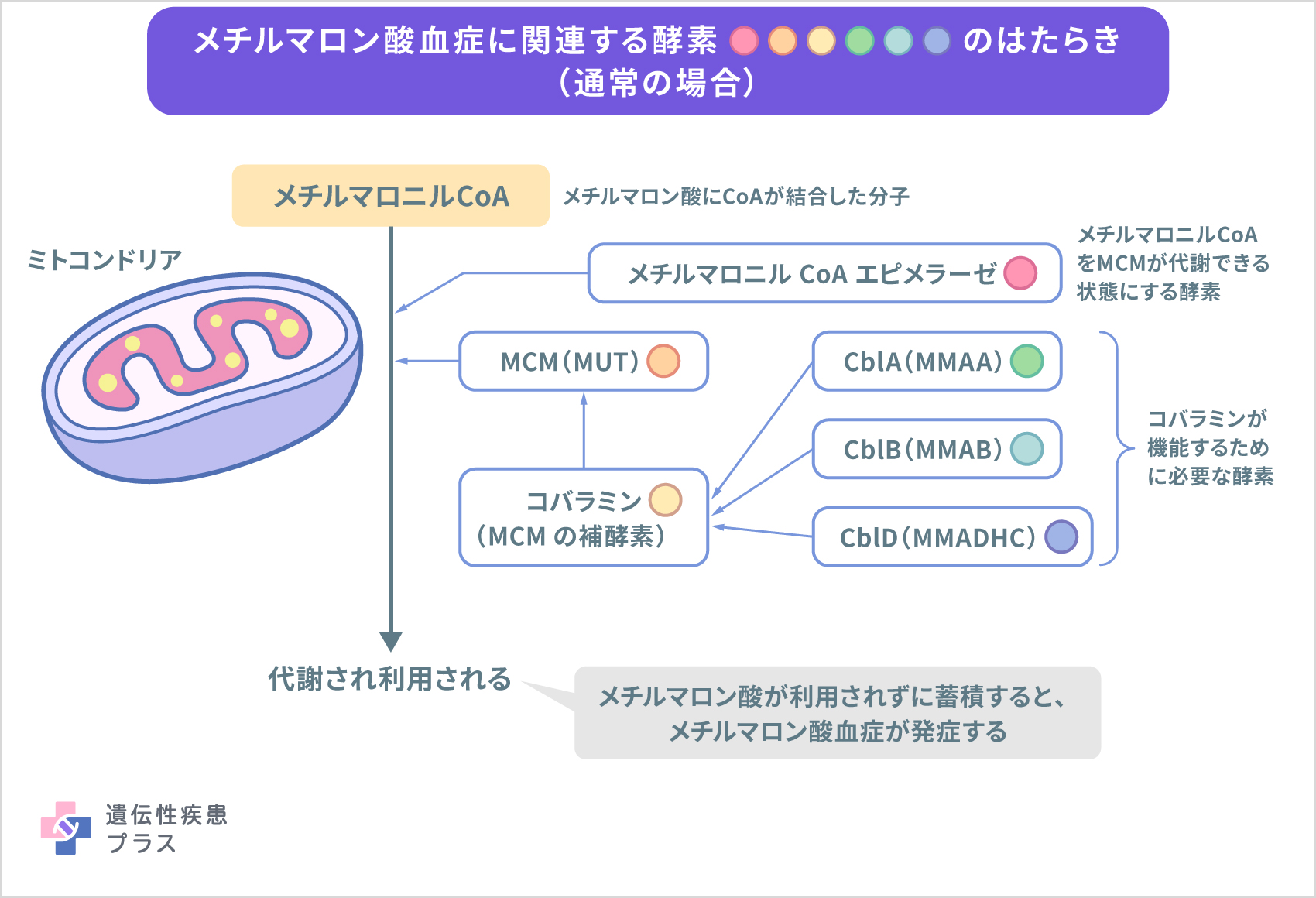
細胞内小器官ミトコンドリアにおいて、メチルマロン酸に補酵素A(CoA)が結合したメチルマロニルCoAは、MCMと補酵素であるコバラミンによって代謝される。メチルマロニルCoAは、メチルマロニル CoA エピメラーゼによってMCMによる代謝が可能な状態に変換される必要がある。また、コバラミンの産生にはCblA、CblB、CblDなどの酵素が必要。青枠内にメチルマロン酸血症に関連する酵素を示した。
メチルマロン酸血症は上記のいずれの遺伝子変異も常染色体劣性(潜性)遺伝形式で遺伝します。常染色体劣性(潜性)遺伝形式は、両親がともに遺伝子の片方に変異を持つ(保因者)場合、子どもは4分の1の確率で発症します。また、2分の1の確率で保因者となり、4分の1の確率でこの遺伝子の変異を持たずに生まれます。

どのように診断されるの?
難病情報センターではメチルマロン酸血症について以下の診断基準が示されています。
<メチルマロン酸血症の診断>
下記①に加えて、②の特異的所見があり、③でビタミンB12欠乏症とホモシスチン尿症を除外できればメチルマロン酸血漿の診断が確定します。
①血中アシルカルニチン分析:プロピオニルカルニチンの増加が見られる。
②尿中有機酸分析:メチルクエン酸、3-ヒドロキシプロピオン酸、3-ヒドロキシグリシンなどの排泄増加が見られる。これらの有機酸はプロピオン酸血症と共通の所見であるが、メチルマロン酸血症ではメチルマロン酸の排泄増加を認める。
③ビタミンB12欠乏症とホモシスチン尿症の除外:血漿総ホモシステイン濃度:正常(<15μmol/L)、血清ビタミンB12:正常(>200pmol/L)
上記によってビタミンB12欠乏症とホモシスチン尿症でないことを確認する。
<病型の確定>
メチルマロン酸血症の原因となっているMCM欠損症およびコバラミン代謝障害(cblA、cblB、cblD)の確定には下記の酵素活性測定や遺伝子解析が必要となる。
酵素活性検査:MCMの酵素活性を測定して、低下が認められればMUT遺伝子の異常によるMCM欠損症と診断される。
遺伝子解析:MCM欠損症についてはMUT遺伝子、cblAはMMAA遺伝子、cblBはMMAB遺伝子、cblDはMMADHC遺伝子の変異を確認する。遺伝子診断は化学診断・酵素診断によって目標を絞り込んだ上で最終確認として実施する。
<新生児マススクリーニング>
新生児マススクリーニングで、プロピオニルカルニチン(C3)の増加およびC3のアセチルカルニチン(C2)に対する比C3/C2の上昇を認めた無症状例は、メチルマロン酸血症またはプロピオン酸血症に罹患している可能性がある。
一般検査(末梢血・一般生化学検査)に加え、血糖、血液ガス、アンモニア、乳酸、血中ケトン体分画、血清ビタミンB12濃度、血清総ホモシステイン、メチオニン濃度を測定し、尿中有機酸分析、酵素活性、遺伝子解析による確定診断を行う。
どのような治療が行われるの?
メチルマロン酸血症の治療はその原因により異なります。コバラミンはMCMの作用を助ける役割をしているため、コバラミンが特効薬となるタイプが知られています。しかし、コバラミンが効果を示さない場合は、現時点で原因となる酵素の異常を回復させる治療法はなく、対症療法が主となります。重症の場合は、タンパク質摂取を中止し、ブドウ糖投与およびアシドーシスに対してアルカリ溶液の点滴を行います。アンモニアという毒性物質が上昇する場合があり、この場合はアンモニアに対する解毒剤が投与されます。また、カルニチンというメチルマロン酸と結合してこれらを排泄させる薬剤を投与します。これでも改善が乏しい場合には血液透析が行われます。一旦、回復した場合、栄養を十分投与するために夜間は経鼻チューブ(もしくは胃に直接穴をあけた胃ろうチューブ)という胃にいれた管から専用の栄養剤を数時間かけて点滴のように補給します。また、早期発症の重症例に対しては生体肝移植も考慮されます。生体肝臓移植を実施することで発作を起こしにくくなり、入院回数も少なくなることが報告されています。しかし、肝臓移植後もゆっくりと神経の障害と腎臓の障害が進むことが治療上の課題となっています。
どこで検査や治療が受けられるの?
日本でメチルマロン酸血症の診療を行っていることを公開している、主な施設は以下です。
- 東北大学病院 小児科
- 千葉県こども病院 代謝科
- 東京慈恵会医科大学附属病院 代謝外来
- 国立成育医療研究センター
- 浜松医科大学医学部附属病院 小児科
- 藤田医科大学医学部小児科学 代謝外来
- 島根大学医学部 小児科
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
メチルマロン酸血症の患者会で、ホームページを公開しているところは、以下です。
参考サイト
- 難病情報センター
- 小児慢性特定疾病情報センター メチルマロン酸血症
- MedlinePlus
- 日本先天代謝異常学会、新生児マススクリーニング対象疾患等 診療ガイドライン2019
- KEGG DISEASE:メチルマロン酸尿症






