



どのような病気?
脊髄小脳変性症(SCD)は、小脳を中心とした神経の変性によって、主に運動失調症状(小脳性運動失調、小脳失調)をきたす病気の総称です。後頭部の下側に位置する小脳に変性があると、複数の筋肉をバランスよく協調させて動かすことができなくなり、運動失調症状が起こります。具体的には、箸を使う・字を書くなどの細かい動きがしにくい、歩く際にふらつく、手が震える、ろれつが回らない、などで、こうした症状がゆっくりと進行します。「変性」とは、原因が不明な病気、つまり、感染症、中毒、腫瘍、栄養素の欠乏、奇形、血管障害、自己免疫性疾患などはっきりと原因が特定されるもの以外のさまざまな原因による病気を指します。SCDには、変性が脊髄、大脳、脳幹、末梢神経に及ぶものも含まれています。また、SCDには、運動失調症状のほか、パーキンソニズム(パーキンソン病様の症状)、錐体路障害、末梢神経障害、認知症など、さまざまな症状を呈する病気が含まれています。
多系統萎縮症(MSA)という、国の指定難病制度上はSCDと区別されている関連疾患があります。MSAは、もともと別々の病気とされていた、オリーブ橋小脳萎縮症、線条体黒質変性症、シャイ・ドレーガー症候群の3つが病型の異なる同じ病気と判明し、まとめて名付けられたものです。オリーブ橋小脳萎縮症の主症状は運動失調症状で、まさにSCDに当てはまります。しかし、線条体黒質変性症の主症状はパーキンソニズム、シャイ・ドレーガー症候群の主症状は自律神経症状であることから、国の指定難病としては、MSA(指定難病17)はSCD(指定難病18)と別に認定されています。また、主に足の突っ張り、歩行障害などの症状が起こる神経変性疾患の「痙性対まひ」は、SCDに当てはまるものが含まれるため、指定難病18の対象疾患となっています。また、SCDは小児慢性特定疾病の対象疾患にもなっています。
現在ではSCDに含まれる病気のうち、多くのものでその原因がわかってきています。例えば、症状から、当初SCDと診断されていても、遺伝的な検査の結果、ミトコンドリア病やプリオン病だと判明した場合には、そちらに病名を変更することになります。しかし、一部まだ原因の解明されていない病気も残されており、これらの病気の解明に向けて、患者さんの協力のもとで、研究が進められています。
その他、反復発作性運動失調症という、遺伝子変異が原因で運動失調症が繰り返し発作的に現れる病気があります。これは、SCDと同じ遺伝子の変異で生じたり、小脳の萎縮が見られたり、わずかに進行したり、発作が起きていない時期にも症状が見られる病型があったりすることから、現在のところSCDとして考えるのが妥当とさています。一方で、何らかの原因で小脳の形成不全がある小脳低形成は、現在SCDには含められていません。
SCDの発症年齢や予後は、疾患の種類によりさまざまです。日本にSCDの患者さんは、MSAを含めると約4万人いると推定されています。令和4年度医療受給者証保持者数によると、SCDは2万6,476人、MSAは1万808人でした。また、SCDのうち、痙性対まひは5%程度を占めています。
SCDには遺伝性の病気と孤発性の病気が含まれています。ここで言う「孤発性」とは、家族歴がみられない場合を指します。具体的には、「両親、兄弟姉妹、子ども」(1度近親者)、および、「祖父母、叔父・叔母、おい・めい、孫」(2度近親者)内に似た症状の病気の人がおらず、さらに、両親が血族結婚をしておらず、命に関わる病気等で命を落としていない限り、両親ともに60歳以上生存している場合です。
日本のSCD患者さんの約3分の1は遺伝性、約3分の2(67.2%)は孤発性とされています。孤発性SCDのほとんどはMSAまたは皮質性小脳萎縮症(CCA)と診断されており、これらはともに青年期以降に発症します。孤発性の場合、生活習慣や食習慣との間に、今のところ明らかな関係は見られず、病気の進行を左右するような食習慣などもわかっていません。また、孤発性SCDは、家族歴がはっきりせず一見孤発性に見えている遺伝性のSCDや、他の病気からの二次性の小脳萎縮症などと、慎重に鑑別されます。
遺伝性SCDは、それぞれ遺伝子別に番号が付けられています。そのうち、日本人に多いのは、脊髄小脳失調症3型(SCA3、以下同様に略)、SCA6、SCA31、歯状核赤核淡蒼球ルイ体萎縮症(DRPLA)です。このうち日本で最も頻度の高い遺伝性SCDのSCA3はマチャド・ジョセフ病とも呼ばれており、小脳失調に加え、足の突っ張り、パーキンソニズム、末梢神経障害、目の症状などをきたすことがあります。SCA3の発症年齢は、10代での若年発症から70代での高齢発症までさまざまです。発症年齢により特徴が異なるため、年代別にさらにI~IV型に分類されています。SCA6は、日本で2番目に多い遺伝性SCDで、西日本に多い傾向があります。発症年齢は中年以降と比較的高齢です。SCA31は、日本人特有とされている遺伝性SCDで、多くが50代以降に発症します。DRPLAは、子どもで発症する人から高齢で発症する人までさまざまで、発症年齢によって症状が異なります。子どものSCDで多いのは、「眼球運動失行と低アルブミン血症を伴う早発型失調症(EAOH/AOA1)」です。子どもで発症する遺伝性SCDとして、欧米ではフリードライヒ運動失調症が高頻度とされていますが、日本でこの病気の症例報告はまだありません。
何の遺伝子が原因となるの?
SCDの約3分の1を占める遺伝性SCDは、その原因遺伝子によって、常染色体優性(顕性)遺伝形式、常染色体劣性(潜性)遺伝形式、X連鎖性遺伝形式、ミトコンドリア遺伝など、さまざまな遺伝形式で受け継がれます。日本で見られる遺伝性SCDは、ほとんどが常染色体優性(顕性)遺伝形式をとるものです。
常染色体優性(顕性)遺伝性SCD(AD-SCD)は、国際ヒトゲノム解析機構HUGOにより、脊髄小脳失調症の略語SCAの後ろに番号を付けた形で登録されています。番号は登録された順番を示し、重複や欠番もありますが、2021年9月時点でSCA48まで登録されています。米ジョンズ・ホプキンス大学が運営している、ヒトの遺伝性疾患とその遺伝子のオンラインカタログOMIMには、最新の常染色体優性(顕性)SCDの原因遺伝子とその情報のリストが掲載されています。日本で頻度が高いAD-SCDは、SCA3、SCA6、SCA31、DRPLAです。

常染色体劣性(潜性)遺伝性SCD(AR-SCD)は、日本ではまれですが、世界的には多数の疾患が報告されています。AR-SCDは、既に病名が広く定着しているものを除き、SCARの後ろに番号を付けた形で登録されています。2021年9月時点のOMIM検索では、SCAR30までが原因遺伝子とともに登録されています。AR-SCDの代表は、フラタキシン(frataxin、FXN)遺伝子の変異が原因のフリードライヒ運動失調症(FRDA)ですが、日本人での報告はありません。日本で報告のあるAR-SCDの代表は、アプラタキシン(aprataxin、APTX)遺伝子の変異が原因の、「眼球運動失行と低アルブミン血症を伴う早発型失調症(EAOH/AOA1)」です。サクシン(sacsin、SACS)遺伝子の変異が原因のシャルルヴォア・サグネ型常染色体劣性(潜性)遺伝性痙性失調症(ARSACS)は、カナダの一地域で多く見られる疾患ですが、日本でも報告されています。この病気は、複合型痙性対まひに分類されることもあります。そのほか、TTPA遺伝子、ATM遺伝子、PNPLA6遺伝子の変異によるAR-SCDなども日本で報告されています。

X連鎖性SCDは主に、脆弱X関連振戦/失調症候群(FXTAS)、X連鎖性脊髄小脳失調症1型(SCAX1)、X連鎖性脊髄小脳失調症5型(SCAX5)です。このうち、FXTASは日本からも報告されています。FXTASとSCAX1は進行性の経過をとりますが、SCAX5は非進行性です。患者さんは主に男性ですが、女性の報告もあります。OMIM登録情報によると、FMR1遺伝子が原因となるFXTASはX連鎖優性(顕性)遺伝形式、ATP2B3遺伝子が原因となるSCAX1とXq25-q27.1領域が原因となるSCAX5は、X連鎖劣性(潜性)遺伝形式とされています。
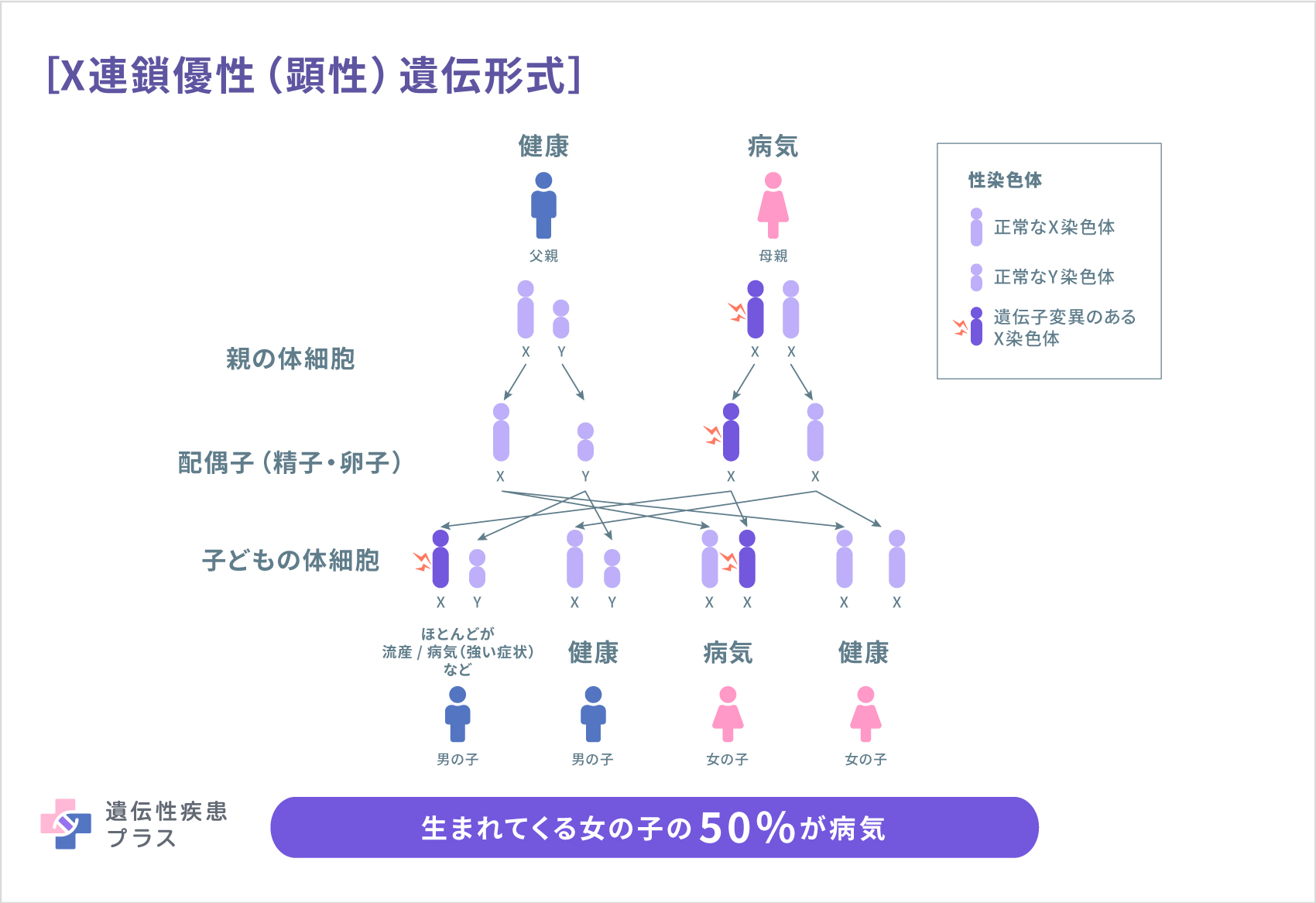
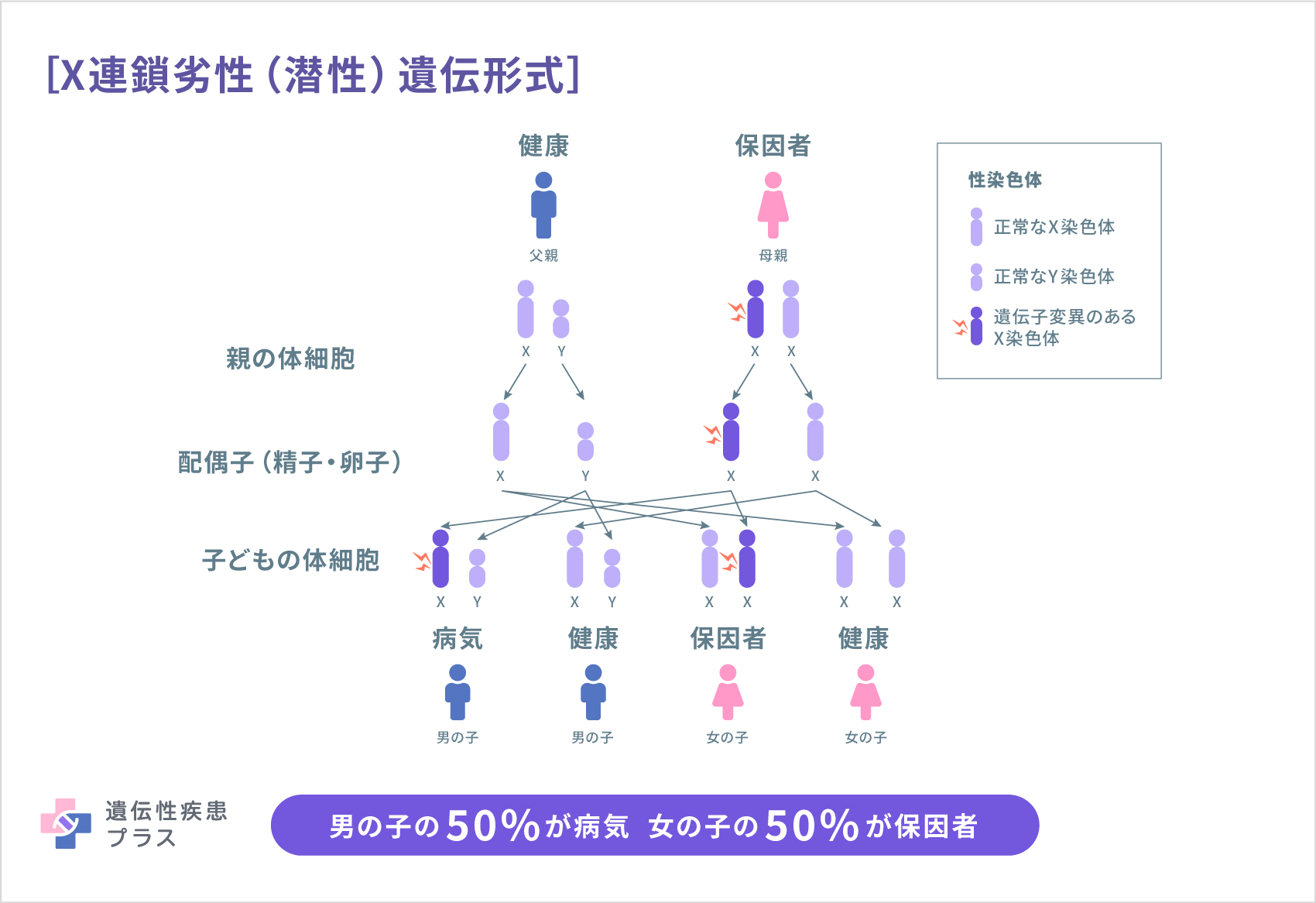
遺伝性痙性対まひ(HSP)も、たくさんの原因遺伝子が見つかっています。HSPは、SPGの後ろに番号を付けた形で登録されています。2021年9月時点のOMIM検索では、SPG83までが原因遺伝子とともに登録されています。HSPも、常染色体優性(顕性)遺伝、常染色体劣性(潜性)遺伝、X連鎖性遺伝、ミトコンドリア遺伝と、さまざまな遺伝形式のものがあります。頻度的には、常染色体優性(顕性)遺伝性の病気が多く、常染色体劣性遺伝(潜性)性は少なく、X連鎖性とミトコンドリア遺伝性はまれです。日本では、常染色体優性(顕性)遺伝性の中でSPG4が最も多く、SPG3A、SPG31、SPG10、SPG8と続きます。常染色体劣性(潜性)遺伝性で多いのはSPG11です。
ミトコンドリア異常によるSCDにおけるミトコンドリア遺伝に関しては、ミトコンドリア病の解説記事をご参照ください。
このように、遺伝性SCDでは、多くの原因遺伝子が判明していますが、その遺伝子の働きや発病のメカニズムについてはまだよくわかっていない部分も多くあります。
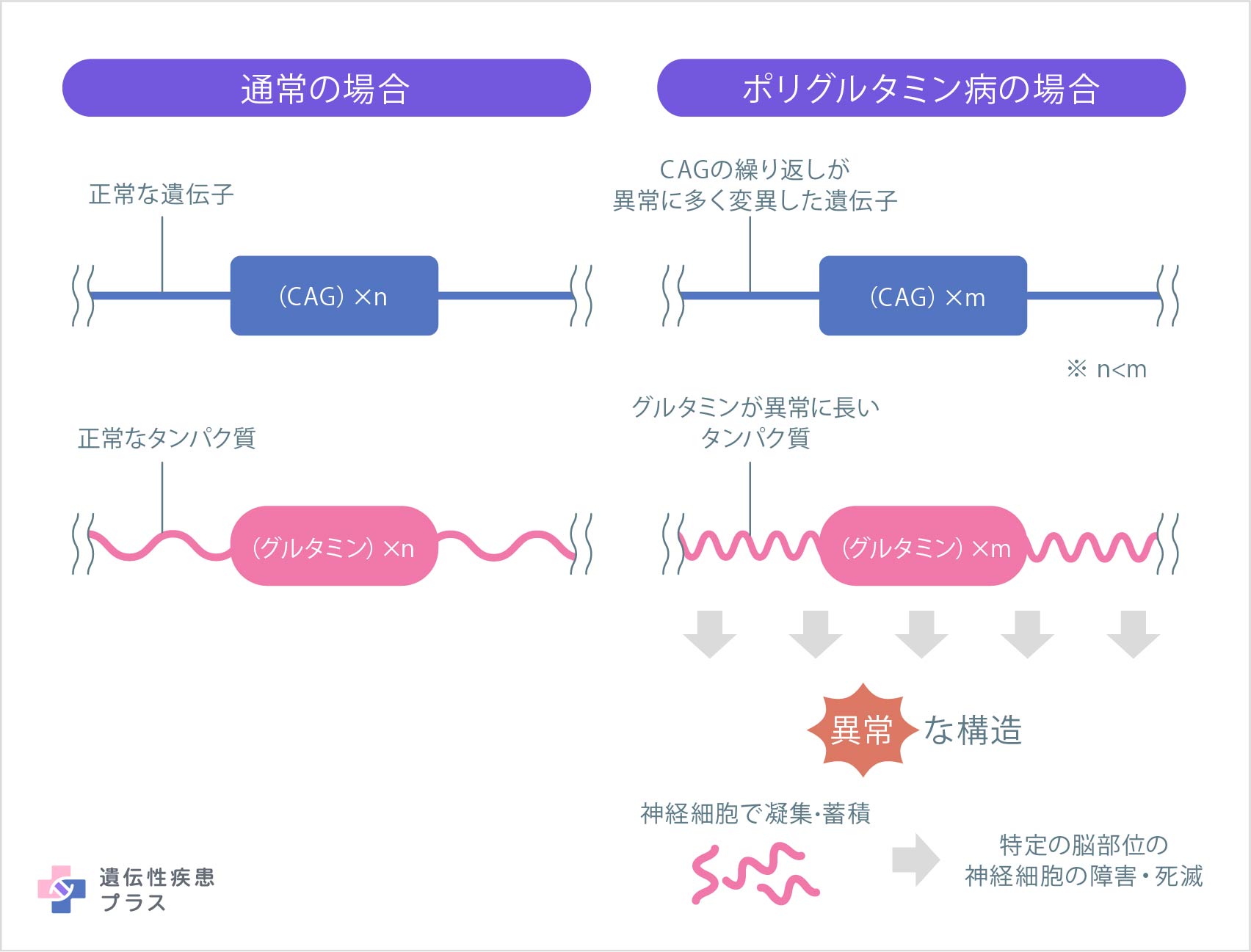
一方、遺伝性SCDの遺伝子変異で多く見られるのは、3~5塩基程度の繰り返し配列が長く起こる変異です。特に、SCA3、6、DRPLAなどに共通して見られるのが、「CAG」というDNA配列(遺伝暗号)の繰り返し(3文字の繰り返しなので「トリプレットリピート」と呼ばれます)が異常に長い状態です。なぜこの繰り返しが長くなるのかは、まだ解明されていませんが、CAGは「グルタミン」というアミノ酸の遺伝暗号になっているため、この繰り返しが多いことで、こうした遺伝子から作られるタンパク質は、グルタミンが異常に長くつながったものになります。このタンパク質が、神経細胞の障害に関連していると考えられています。しかし、その詳細なメカニズムはまだ完全に解明されておらず、発病や進行を阻止できる治療法開発のために、研究が続けられています。ハンチントン病や球脊髄性筋萎縮症も、同様の仕組みで起こっている病気で、これらは「ポリグルタミン病」と名付けられています。
どのように診断されるの?
SCDには、さまざまな病気が含まれていますが、医師による問診や診察、検査を経てSCDと診断されます。問診では病歴などが聞かれます。診察では、小脳失調症状の確認などが行われます。脳のMRIやCT検査は、小脳の萎縮、脳幹部や大脳の異常の有無について調べるために行われます。脳の血流状態を評価する検査や、末梢神経障害の有無について調べる検査が行われることもあります。自律神経障害の評価は、横になった状態から立った状態になったときに血圧の変化を測定する検査や、排尿障害に関する泌尿器科での検査などで行われます。
SCDの診断基準として、難病情報センターに掲載されている情報は、以下です。
SCDと確定診断(Definite)される場合
SCD・痙性対まひに合致する症候と経過があり、遺伝子診断か神経病理学的診断がなされている場合。
SCDがほぼ確実(Probable)と診断される場合
(1)SCDに合致する症候があり、「小脳性ないしは後索性の運動失調または痙性対まひを主要症候とする」「徐々に発病し、経過は緩徐進行性である」「頭部MRIやX線CTで小脳や脳幹の萎縮が認められる(病型や時期によっては大脳基底核病変や大脳皮質の萎縮などが認められることもある)」を満たす場合で、次の原因による二次性小脳失調症(鑑別疾患※)ではないとされた場合。
※鑑別疾患:脳血管障害、腫瘍、アルコール中毒、ビタミンB1・B12・葉酸欠乏、薬剤性(フェニトインなど)、炎症(神経梅毒、多発性硬化症、傍腫瘍性小脳炎、免疫介在性小脳炎(橋本脳症、シェーグレン症候群、グルテン失調症、抗GAD抗体小脳炎))、甲状腺機能低下症、低セルロプラスミン血症、脳腱黄色腫症、ミトコンドリア病、二次性痙性対麻痺(脊柱疾患に伴うミエロパチー、脊髄の占拠性病変に伴うミエロパチー、多発性硬化症、視神経脊髄炎、脊髄炎、HTLV-I関連ミエロパチー、アルコール性ミエロパチー、副腎ミエロニューロパチーなど。
(2)SCD・痙性対まひに合致する症候があり、かつその家系内の他の発症者と同一とみなされる場合(遺伝子診断がなされていない場合も含む)。
SCDの疑い(Possible)と診断される場合
SCD・痙性対まひに合致する症候があり、「小脳性ないしは後索性の運動失調または痙性対まひを主要症候とする」「徐々に発病し、経過は緩徐進行性である」「頭部MRIやX線CTで小脳や脳幹の萎縮が認められる(病型や時期によっては大脳基底核病変や大脳皮質の萎縮などが認められることもある)」を満たす、または、痙性対まひに合致する症候があり、「小脳性ないしは後索性の運動失調または痙性対まひを主要症候とする」「徐々に発病し、経過は緩徐進行性である」を満たすが、上記鑑別疾患が除外できない場合。
その他、診断の際の主要項目として挙げられている臨床的な特徴として、以下が挙げられています。
- 病型によっては遺伝性を示す。その場合、常染色体優性遺伝性であることが多いが、常染色体劣性遺伝性、X染色体劣性遺伝性の場合もある。
- その他の症候として、錐体路症候、パーキンソニズム(振戦、筋強剛、無動)、自律神経症候(排尿困難、発汗障害、起立性低血圧)、末梢神経症候(しびれ感、表在感覚低下、深部覚低下)、高次脳機能障害(非薬剤性の幻覚、失語、失認、肢節運動失行以外の失行)などを示すものがある。
一方、MSAの診断基準として掲載されている情報は、以下です。
MSAと確定診断(Definite)される場合
病理学的に確定診断された場合。
MSAがほぼ確実(Probable)と診断される場合
パーキンソニズム(筋強剛を伴う運動緩慢、振戦もしくは姿勢反射障害)、または小脳症候(歩行失調、小脳性構音障害、小脳性眼球運動障害、四肢運動失調)に、自律神経症候(「MSAの疑い」の基準に満たない程度の起立性低血圧や排尿障害、睡眠時喘鳴、睡眠時無呼吸もしくは勃起不全)を伴い、かつ錐体路徴候が陽性であるか、もしくは画像検査所見(MRIもしくはPET・SPECT)で異常を認める場合。
MSAの疑い(Possible)と診断される場合
レボドパに反応性の乏しいパーキンソニズムもしくは小脳症候のいずれかに明瞭な自律神経障害を呈する(抑制困難な尿失禁、残尿などの排尿力低下、勃起障害、起立後3分以内において収縮期血圧が30mmHgもしくは拡張期血圧が15mmHg以上の下降、のうちの1つを認める)場合。
多くの場合、遺伝性SCDと孤発性SCDの区別は、症状や画像検査からつけることができます。そのため、両者の区別のための遺伝子検査は必ずしも必要ではありません。一方で、皮質性小脳萎縮症など、症状や画像検査だけでは両者の区別が難しい病気も一部あります。正確な病型診断のために、一部の大学病院など専門的な医療機関で遺伝子検査が実施される場合があります。なお、今のところSCDの遺伝子検査は保険適用されていません。
どのような治療が行われるの?
今のところSCDを根本的に治すような治療法は見つかっていません。そのため、それぞれの症状を和らげるための対症療法を中心に治療を受けることになります。
一方で、多くのSCDで病気の原因が判明してきており、その原因に基づいた治療法開発の研究が、世界中の研究者によって進められています。動物実験レベルで、症状の進行を妨げる薬剤の候補も多数見つかってきています。その中には遺伝子治療や核酸医薬の候補も含まれています。こうした薬や治療法の候補がヒトで安全・有効であるかを確かめ、治療として確立させていくためには、多くの患者さんの協力が不可欠であることが、難病情報センターのサイトにも明記されています(この記事の最後に、今国内で行われているSCDの臨床試験情報へのリンクがあります)。
対症療法については、例えば、運動失調に対して、甲状腺刺激ホルモン放出ホルモン(TRH)誘導体のタルチレリン(製品名:セレジスト)という内服薬、TRH製剤の酒石酸プロチレリン(製品名:ヒルトニン)という注射薬が使われています。タルチレリンの作用機序は十分にわかっていませんが、臨床試験で小脳失調に対する効果が確認されており、また、注射薬ではなく内服薬であることもあり、全般に広く使用されています。その他、パーキンソン症状にはパーキンソン病の治療薬L-dopaを中心とした薬による治療、便秘や排尿障害などの膀胱直腸障害については薬によるコントロールや導尿、痙性に対しては抗痙縮薬の使用など、症状に応じた薬で治療が行われます。
日常生活では、立ち上がったときや歩いているとき(特に歩き出したときや向きを変えたとき)に、ふらついて転倒することがあるので、注意しましょう。転倒を防ぐため、廊下、お風呂、トイレなどに、手すりを設置するなど工夫をすると良いでしょう。疾患にもよりますが、運動機能の維持や残存機能の活用のために、バランス訓練や歩行訓練などのリハビリテーションが効果的であり、その効果は終了後しばらく持続することが示されています。リハビリテーションのメニューはその人の状態によって異なるため、主治医や理学療法士と相談して、ご自分に合った方法で進めましょう。
疾患によっては、進行すると食べ物や飲み物が上手に飲み込めなくなる(嚥下障害)ことがありますが、この場合、誤嚥性肺炎のリスクが高まります。食事の際にむせるようなことが続いたら、早めに医師に相談しましょう。また、食べ物を細かく刻む、とろみをつけるなどの対応や、食後の口腔ケアも重要です。必要に応じて、嚥下や発声、呼吸のリハビリテーションが行われる場合もあります。
その他、日常では、その人のできる範囲で安全を確保しながら、立ったり座ったり歩いたり、関節を動かしたりといった訓練を実施していくことが重要です。お酒はふらつきの悪化につながることがあるため、控えめにしましょう。便秘に対しては水分、食物繊維、ヨーグルトや発酵食品の摂取、また、可能な範囲での運動を心掛けましょう。
ケアをするご家族も、患者さんが起き上がったり歩いたり、車椅子に乗ったりするときの介助をする際に、無理な姿勢を取ったり無理な力が入ったりすることで、腰や肩を痛める可能性があります。そのリスクをなるべく抑えるために、こうした介助について、看護師や理学療法士からアドバイスを受けておきましょう。
SCDと向き合っていく上で大事なことは、1人で悩まずに、周囲の人たちに相談して不安を減らし、できるだけ前向きに生活していくことです。医療機関のほか、国や自治体などには、患者さんの状況に応じたさまざまな制度や支援があります。現在の体の機能と生活の質を少しでも維持できるよう、また、ご家族のケアの負担をできるだけ増やさないよう、主治医、看護師、医療ソーシャルワーカー、ケアマネージャー、自治体の窓口や保健所、難病相談センター、ハローワーク、患者会などに相談するようにしましょう。
どこで検査や治療が受けられるの?
日本でSCDの診療を行っていることを公開している、主な施設は以下です。
- 北海道大学病院
- 札幌医科大学附属病院 脳神経内科
- 仙台西多賀病院 筋神経疾患研究室
- 福島県立医科大学附属病院 脳神経内科
- 新潟大学脳研究所 脳神経内科
- 信州大学医学部医学科内科学第三教室 脳神経内科
- 群馬大学医学部附属病院 脳神経内科
- 千葉大学医学部附属病院 脳神経内科
- 埼玉医科大学総合医療センター 神経内科
- 東京大学医学部附属病院 脳神経内科
- 東京都立神経病院
- 東京科学大学病院 脳神経内科
- 国立精神・神経医療研究センターNCNP病院
- 横浜市立大学附属病院 脳神経内科
- 東海大学医学部付属病院脳神経内科
- 山梨大学医学部附属病院
- 名古屋大学医学部附属病院 脳神経内科
- 森之宮病院 脳神経内科・リハビリテーション科
- 岡山大学病院 脳神経内科
- 鳥取大学医学部附属病院
- 九州大学病院 ブレインセンター
- 鹿児島大学病院 脳神経内科
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
SCDの患者会で、ホームページを公開しているところは、以下です。
参考サイト
- 難病情報センター
- 小児慢性特定疾病情報センター 脊髄小脳変性症
- e-Stat 衛生行政報告例/令和元年度衛生行政報告例 統計表 年度報
- 日本神経学会 脊髄小脳変性症・多系統萎縮症診療ガイドライン2018
- 兵庫県難病相談センター 脊髄小脳変性症
- Online Mendelian Inheritance in Man(R) (OMIM(R))
参考文献:医学書院 医学大辞典 第2版






