



筋ジストロフィーは、筋肉の機能に必須なタンパク質の設計図となる遺伝子の変異により、病理学的な異常を背景として起こる進行性の疾患の総称です。中でも、デュシェンヌ型筋ジストロフィー(DMD)は、小児期に発症する遺伝性筋疾患のうち最も頻度が高く、その遺伝形式から発症者はほとんどが男性です。また、DMDは筋ジストロフィーの中でも特に重症度が高く、原因遺伝子は1つですが、その遺伝子が巨大なため、発症メカニズムは非常に複雑です。一方で、近年、DMDに対する核酸医薬品や遺伝子治療薬など、新しい治療法が開発されてきており、注目を集めています。そこで、筋ジストロフィーとその最新治療について、同疾患の先端治療法開発の第一人者で日本の研究班を長年にわたって組織して来られた、国立精神・神経医療研究センター顧問 (名誉所長)の武田伸一先生に、詳しくお話を伺いました。

筋ジストロフィーとはどのような病気でしょうか
筋ジストロフィーの定義として、歴史的に次の3つの要素が挙げられます。
- 患者さんの様子から「進行性の筋力低下を呈する疾患」とされてきた
- その患者さんの筋肉組織を病理学的に調べてみると、「変性と再生を繰り返す」状態が見られる
- 筋肉の機能に必須なタンパク質をコードする遺伝子に「変異」がある
筋肉は、「筋線維」が単位となって構成されています。なぜ細胞と言わずに筋線維と言うのかというと、細胞が融合して多核(核がたくさん存在する状態)になっているからです。この筋線維は、3. の遺伝子変異が原因で傷害を受けると、段々と壊れますが、その壊れていく状態を「変性」と言います。筋線維は変性すると、その一部が傷害されますが、傷害されない部分も残り、それが増殖した筋衛星細胞と融合して再生します。これは、多核ならではの仕組みです。しかし再生後、多くの筋線維は再び変性するため、変性と再生を繰り返すことになります。繰り返しの中で、やがて再生が追いつかなくなり、筋線維は消失し、脂肪や線維化に置き換えられていきます。筋ジストロフィーでは、こうした仕組みにより、進行性の筋力低下が起こってきます。
筋肉の機能に必須なタンパク質は複数あり、筋ジストロフィーは、それらのタンパク質を作る遺伝子が変異したことにより起こる病気の総称であるため、多くの病型が含まれます。成人患者さんで最も多いのは筋強直性(きんきょうちょくせい)ジストロフィー、小児患者さんで一番多いのはデュシェンヌ型(Duchenne)筋ジストロフィー(DMD)です。また、DMDと同じ遺伝子の変異で起こる、良性のベッカー型(Becker)筋ジストロフィーもあります。それ以外は、かなり患者数が少なくなってきますが、肢帯型(LG)、顔面肩甲上腕型(FSHD)、眼咽頭型(Oculopharyngeal)、先天性(Congenital)、遠位型(Distal)、エメリー・ドレイフス型(Emery-Dreifuss)などの病型があります。先天性筋ジストロフィーのうち、日本で一番多いのは福山型(FCMD)です。福山型は日本に固有の病型で、小児ではDMDの次に多い病型です。つまり日本では、大人に多い筋強直性、子どもに多いDMDと福山型が代表的な筋ジストロフィーと考えて頂ければと思います。これらの筋ジストロフィーは、新生変異としてお子さんで初めて発症する場合もあるので、必ずしも遺伝性とは言えませんが、全て遺伝子変異が原因となって発症します。
ところで今日お話しする最新治療については、主にDMDに関する話になります。というのは、DMDは見出されてから歴史が長く、また、お子さんが発症し深刻な経過をたどるため、多くの人が注目し、研究も多くなされてきたからです。
DMDについてもっと詳しく教えてください
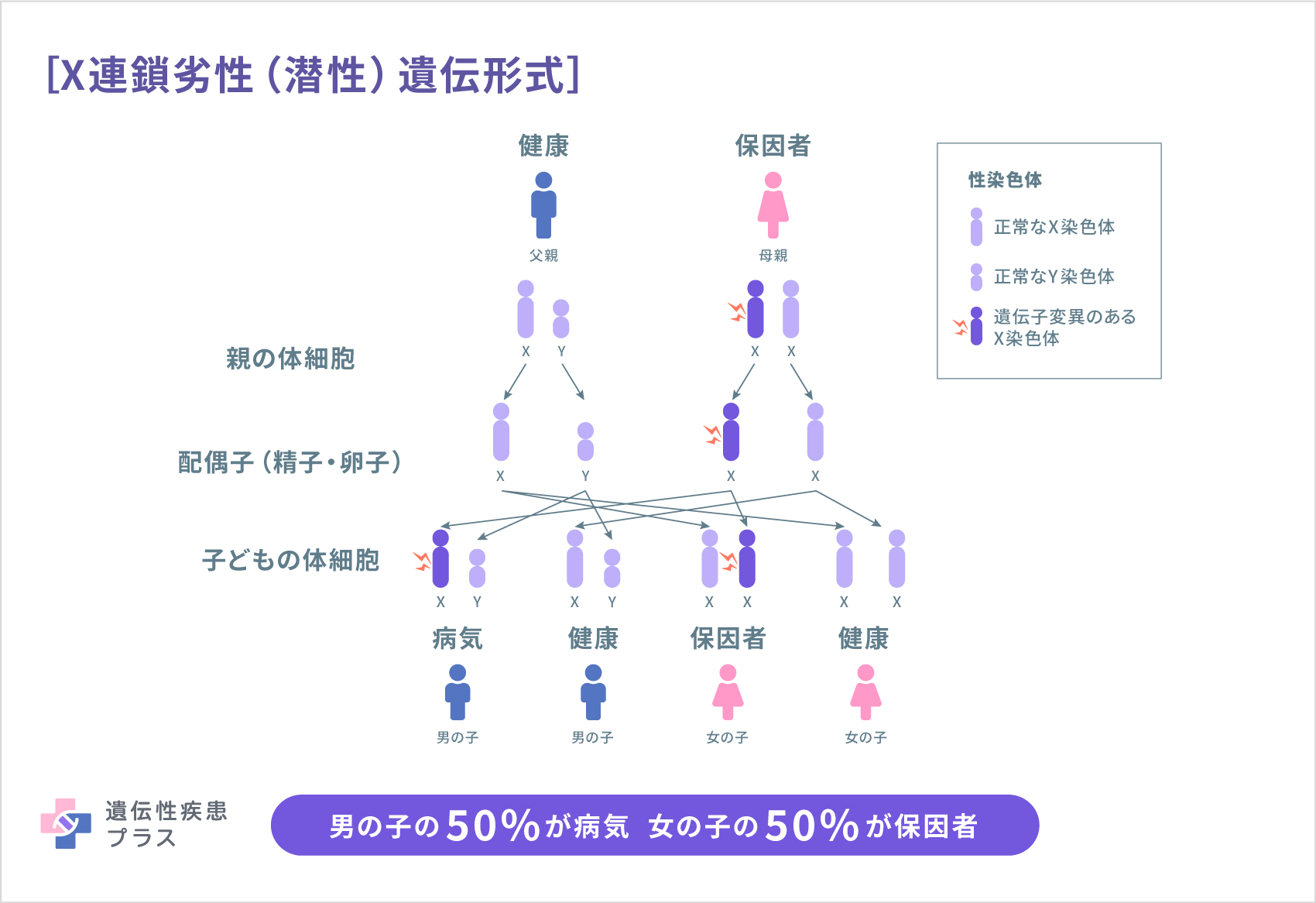
DMDは、小児期に発症する遺伝性筋疾患の中で最も頻度が高く、出生男児約5,000人に1人の割合で発症します。2~5歳に歩行障害で発症後、筋力低下が段々進み、11~13歳で歩行が困難になり、20~40歳くらいで心不全(拡張型心筋症)や呼吸不全で亡くなられます。X連鎖性劣性(潜性)遺伝形式のため患者さんのほとんどが男性です。患者さんのうち、母親が保因者である割合は3分の2で、残りの3分の1は新生変異です。
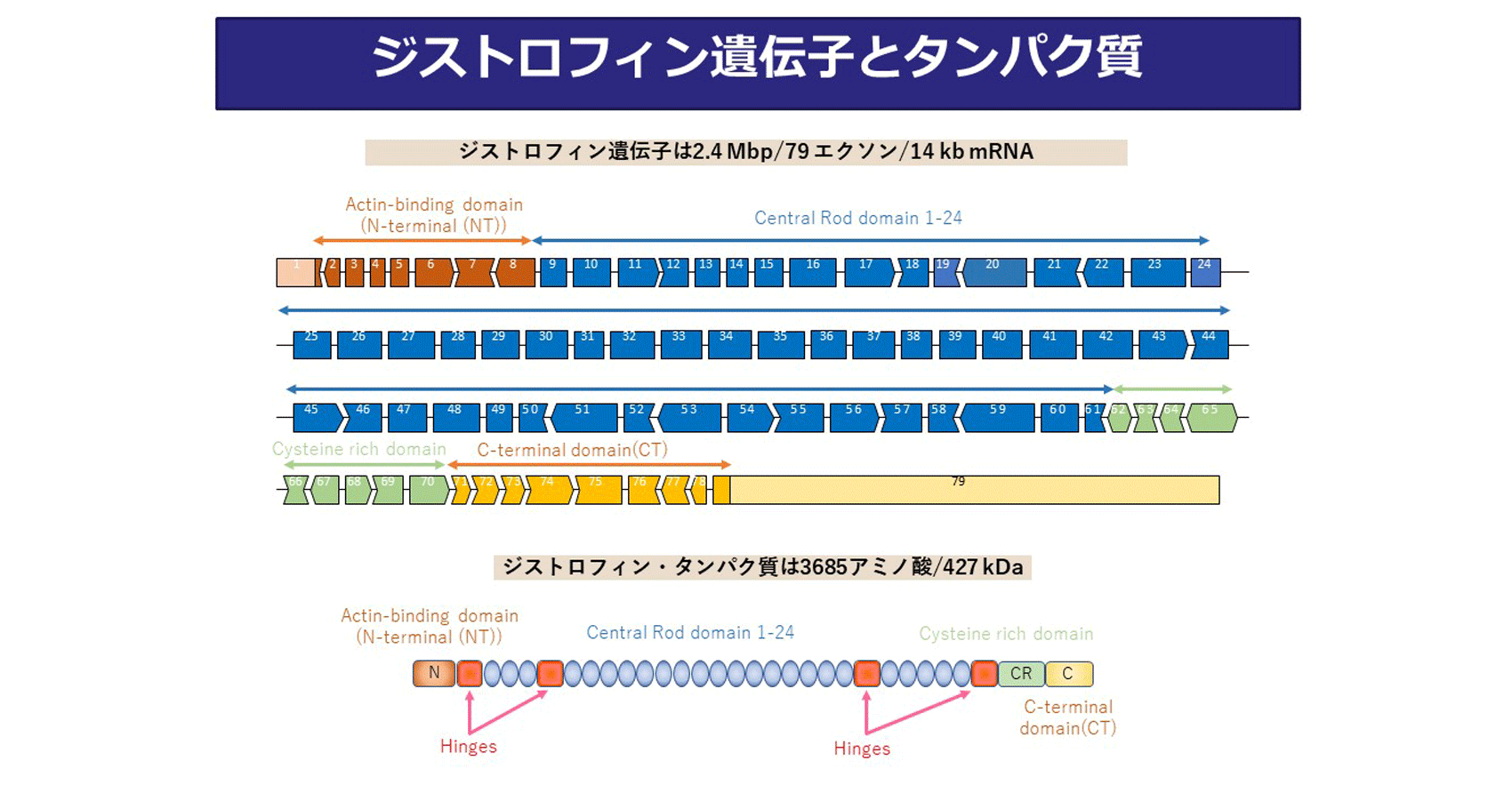
DMDは、「ジストロフィン遺伝子」という巨大な遺伝子の変異で起こります。その大きさは具体的に、遺伝子の長さが2.4 Mbp(メガベースペア、240万塩基対)、エクソンは1~79番目まであり、メッセンジャーRNA(mRNA)の長さは14 kb (キロベース、1万4,000塩基)です。このゲノム遺伝子を基にして作られるジストロフィン・タンパク質も、非常に大きなタンパク質(3,685のアミノ酸から成り、質量は427 kDa)で、細胞膜を内側から裏打ち(裏側から補強)するような位置にあります。ジストロフィンは4つの部分(ドメイン)から成り、まず両端が「N末端ドメイン」と「C末端ドメイン」です。N末端ドメインは、骨格筋の構造と機能に重要なアクチンというタンパク質に結合する部位です。C末端ドメインの内側には「システイン(Cys)リッチ・ドメイン」があり、このCysリッチ・ドメインとC末端ドメインとで、他の複数の分子と結合します。N末端ドメインとCysリッチ・ドメインの間をつなぐところが「ロッド・ドメイン」で、これは24回の繰り返し構造から成っています。ここでまた遺伝子に戻って見ると、エクソン1~8が「N末端ドメイン」、エクソン9~61が「ロッド・ドメイン」、エクソン62~70が「Cysリッチ・ドメイン」、エクソン71~79がC末端ドメインをコードしていることがわかります。
DMDは、ジストロフィン遺伝子の変異により、ジストロフィン・タンパク質が「完全に欠損」するために起こります。ジストロフィンが欠損すると、ジストロフィンに結合するタンパク質もまた、二次的に無くなります。その結果、筋線維の変性・壊死が起こります。そうすると、再生を起こそうとして、免疫細胞(マクロファージや白血球)が浸潤してきます。これらがサイトカインを出し、T細胞が押し寄せてきます。こうして浸潤してきた細胞たちの働きにより、再生が進みます。しかし、最終的には再生は変性を上回ることができず、筋肉は線維化や脂肪化を起こしていきます。
DMD患者さんでは、エクソン単位での欠失変異が多く、エクソン9~61、中でも特にエクソン45~55に変異箇所が集中しています。つまり、このように非常に大きなゲノム遺伝子ですが、変異は均等に起こるのではなく、特定の場所に偏って起こります。これは、日本人でも外国人でも同じ傾向です。国内にRemudy(レムディ)と呼ばれるDMDの患者登録制度があり、DMDの登録者は2,100人を超えていますが、Remudyで調査した結果でも、やはりエクソン45~55に変異が集中していました。こうした理由から、後でお話しする「エクソン・スキップ治療」は、エクソン45~55の範囲をターゲットとして、まずは進められてきています。
DMDではどのような治療が行われるのでしょうか
治療の方向は2つあります。1つは、ジストロフィンの欠損を補う治療です。その方法として、エクソン・スキップや遺伝子治療が挙げられます。ただ、こうした治療では、ジストロフィン遺伝子が非常に大きいことが問題となっています。もう1つは、ジストロフィンの欠損によって起こる不具合を制御する治療です。例えば、筋線維の変性、線維化、脂肪化を何とかして抑えたり、再生を活発にしたりする治療です。
DMDに対する治療アプローチ(研究中のものも含む)
1)ジストロフィン発現の回復を目指すもの
- エクソン・スキップ
- ストップコドン・リードスルー
- ウイルスベクターによる遺伝子治療
- iPS 細胞移植
- ゲノム編集技術の応用
2)筋保護、筋増量、またはジストロフィン欠損を代償する機構の活性化を目指すもの
- 抗炎症剤(例:ステロイド、Vamorolone、Edasalonexent)
- 抗酸化剤(例:Puldysa(Idebenone))
- フォリスタチン増強(例:Givinostat)
- カルシウム代謝(例:Rimeporide)
- 線維化抑制(例:Pamrevlumab)
DMDのエクソン・スキップ治療について教えてください
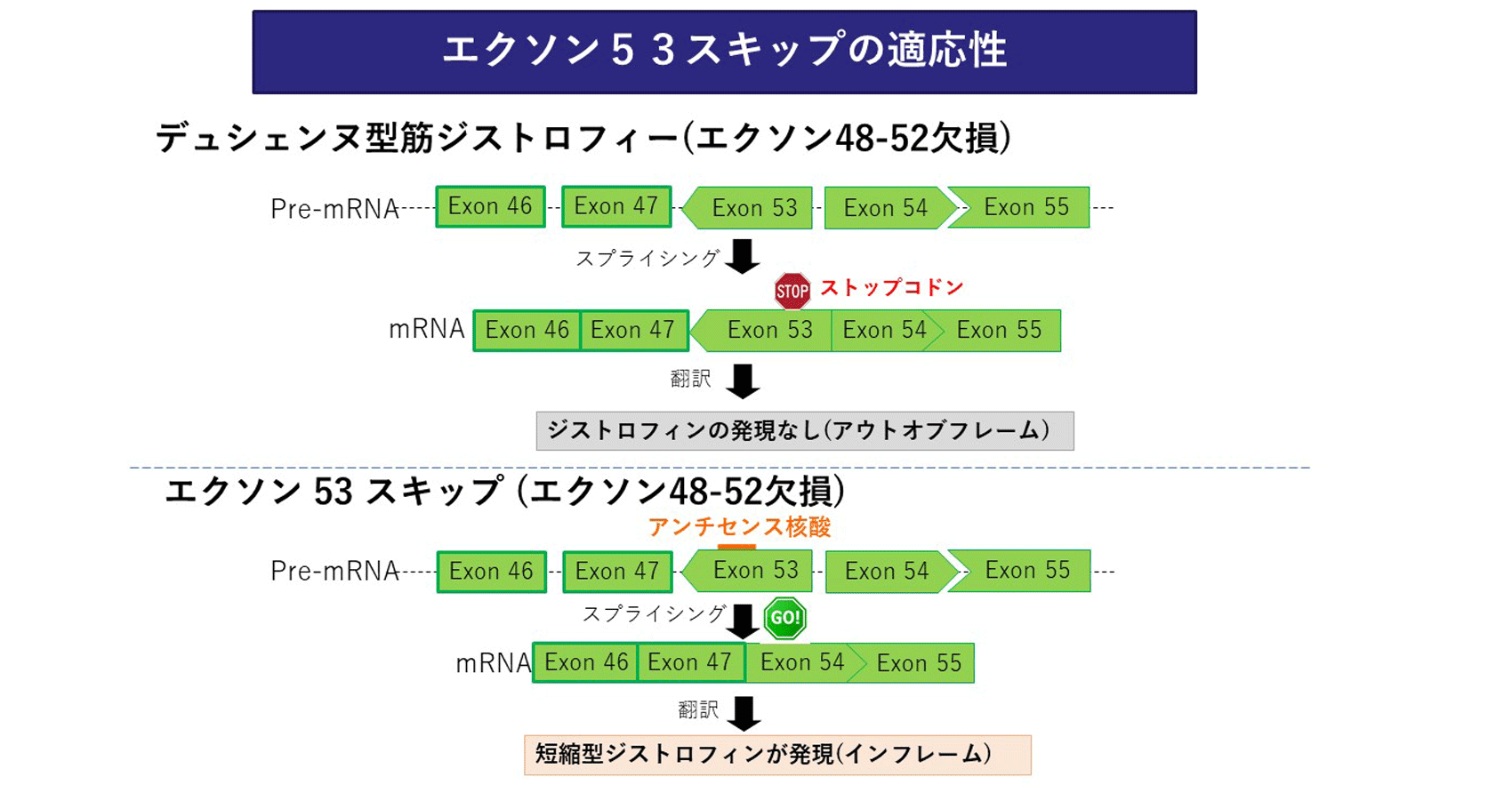
DMDの65%くらいは「欠失」と呼ばれる変異により起こります。例えばエクソン48~52が欠失している患者さんの場合、「…エクソン46、47、(飛んで)、53、54、55…」と続いていきます。通常なら、mRNA上では3塩基が1組でアミノ酸への変換コード(コドン)となっており、これが継続することでアミノ酸がつながったタンパク質が作られていくのですが、ここに欠失があるために、その読み枠がずれてストップ・コドン(タンパク質合成終了のサイン)が生じ、それ以上翻訳が進まなくなります。これをアウト・オブ・フレーム変異と呼びます。このとき、正常な遺伝子を「補う」遺伝子治療も考えられますが、この「足し算」の治療に対し、「引き算」の治療がエクソン・スキップ治療です。この患者さんの場合、スプライシングという機構に働きかけるアンチセンス核酸医薬品を投与することで、欠失直後のエクソン53を飛ばしてやります(スキップします)。そうすると、「…エクソン46、47、(エクソン53まで飛んで)、54、55…」となり、3塩基ずつ読み方のずれが是正されて、ストップ・コドンが生じず、その先までタンパク質の合成が進行していきます(これを、専門的には、アウト・オブ・フレームがイン・フレームになったといいます)。
ただ、作られるようになった短縮型のジストロフィンが、体内で本当に機能するのかどうかが問題です。DMDではジストロフィンが完全欠失する変異ですが、ベッカー型筋ジストロフィー(BMD)では、短縮型のジストロフィンが作られる変異(インフレームな変異)が生じています。BMDの患者さんのジストロフィンは、生まれたときから短いので、体の中で適応できており、16歳を過ぎても歩けますが、治療によりベッカー型と同じような変異ができたときに、どれくらい効果が得られるかは、別問題です。そこで、動物実験や臨床試験で、効果を確認する必要があります。実際、私たちの研究グループはエクソン53スキップ薬「ビルトラルセン」(Viltolarsen、販売名:ビルテプソ点滴静注250mg)開発の過程で、モデル動物を使って、短縮型のジストロフィンが機能することを確認しました。全く筋膜にジストロフィンが確認できなかったモデル動物にエクソン・スキップ治療をしたところ、少し短いジストロフィンが筋膜に回復していることが確認できたのです。その後、臨床試験を経て、承認に至りました。
ビルトラルセンは、日本と米国で承認されていますが、条件付き早期あるいは迅速承認であったため、現在もさまざまな試験が進行しています。例えば、北米における(第2相後の)継続投与試験、プラセボをコントロールにおいた第3相グローバル(国際共同)治験などが実施されています。また、日本でも、並行して製造販売後調査として、自然歴との比較が行われています。承認時には、歩行への効果が評価されましたが、こうした継続的な試験により、上肢や呼吸機能改善の可能性も出てきました。これに関しては、リハビリとどのように組み合わせるかが非常に大事になってきます。また、心機能改善の可能性も検証されてきています。こうした効果が明らかになれば、患者さんの生活が大きく改善されると期待できます。
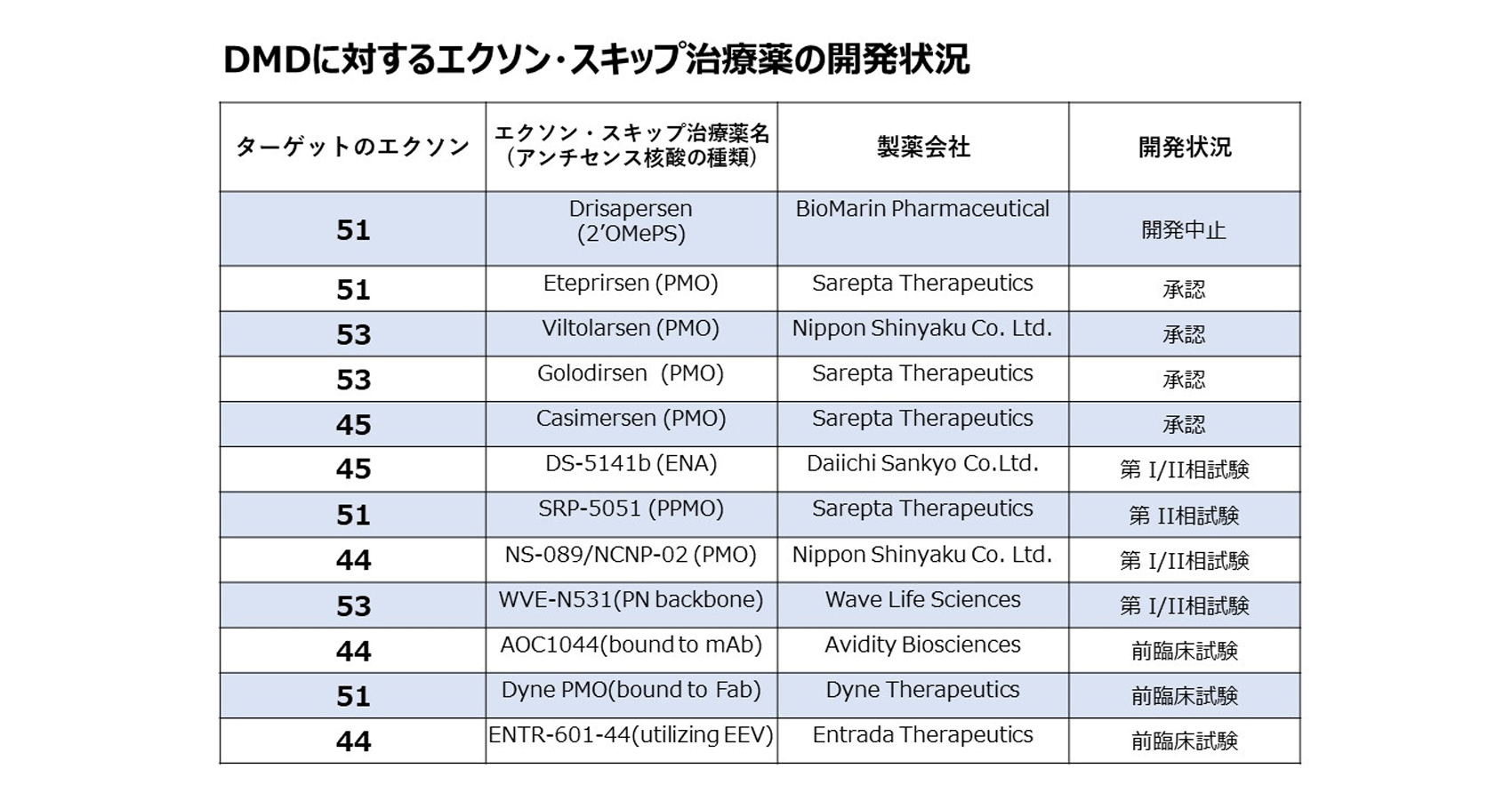
ビルトラルセン以外のエクソン・スキップ治療薬の開発状況を教えてください
DMDに対するその他のエクソン・スキップ治療薬の開発は、特にエクソン51、53、45をターゲットとした治療薬の開発が進んできています。アンチセンス核酸の種類により、それぞれ異なった特徴を持っています。これまでに承認されたものは、PMO(モルフォリノ核酸)という種類で、骨格筋での効果が期待されるものでした。PPMOという種類のアンチセンス核酸は、PMOに、骨格筋に取り込まれやすくなるようなアミノ酸を付けたもので、骨格筋に対し、より強い効果が予想できるほか、ある程度心筋での効果も期待されています。また、第一三共株式会社の進めているENAも、まだなかなか難しい部分もありますが、心筋に対する効果を持つ可能性もあります。これらの新しい薬の治験の進行は、注意深く見守る必要があると思っています。
こうしたエクソン・スキップ治療薬の開発過程では、「n=1試験」が注目されています。DMDは希少性疾患なので、日本国内に患者さんは3,500~4,000人、その中でエクソン53スキップの対象患者さんは10%くらいです。他にもっと患者さんの数が少ない変異もあります。そうした変異に対してエクソン・スキップ治療薬が必要な場合、すでにモルフォリノ核酸は安全性が高いことがわかっているので、例え患者さんが1人であっても治療ができないか、と考え、「安全性の検証が試験管内で示せていれば、患者さんが1人でも治療できるようにしよう」という動きがあります。これがn=1試験と呼ばれるものです。
米国で迅速承認されたDMDの遺伝子治療「ELEVIDYS」について教えてください
遺伝子治療をするには、遺伝子を体内の必要な場所へ運び込む「ベクター」が必要です。ベクターは通常、ウイルスを改変して安全性を高めたもので、骨格筋に対して最も有力なベクターは、「AAVベクターです」。ところがAAVベクターは遺伝子の搭載能力が低く、4.8 kb(1 kbは1,000塩基)までしか載りません。これまでお話ししてきたように、ジストロフィン遺伝子は非常に大きい遺伝子です。いろいろなところを削っても12 kbはあり、とても載りません。
一方、非常に軽症のベッカー型の患者さんの中に、6.3 kbの短いジストロフィン(ミニ・ジストロフィン)を持つ英国の患者さんがいました。私たちはこれを出発点にして、24個あるロッド・ドメイン(繰り返し構造)はどこまで減らすことが可能か、研究を進めました。私たちの研究を手がかりとして、5個のロッド・ドメインを持つミニ・ジストロフィンが米国で治療用に作製されました(下図Δ3990)。これは今、ファイザーが遺伝子治療の開発を進めているものです。その下、4つのロッド・ドメインを持ったもの(ΔR4-23/ΔC)は、マイクロ・ジストロフィンと呼ばれており、これが現在、AAV搭載用として最も主流です。一番下もマイクロ・ジストロフィンで、R16とR17を入れているタイプのものです(μDys-5R)。R16、R17はnNOSという、筋肉の機能維持に重要な血管を拡張する分子の結合部位で、この部分があるとnNOSの発現が回復することを発見した研究グループが、この2つのリピートを残したマイクロ・ジストロフィンを考案しました。ミニ・ジストロフィンもマイクロ・ジストロフィンも、もともと英国の患者さんのミニ・ジストロフィンから出発して、さらに24個あるロッド・ドメインを4~5個に縮めたものです。今のところこれが「短いけれど機能するジストロフィン」としては限界で、これ以上短くは出来ていません。
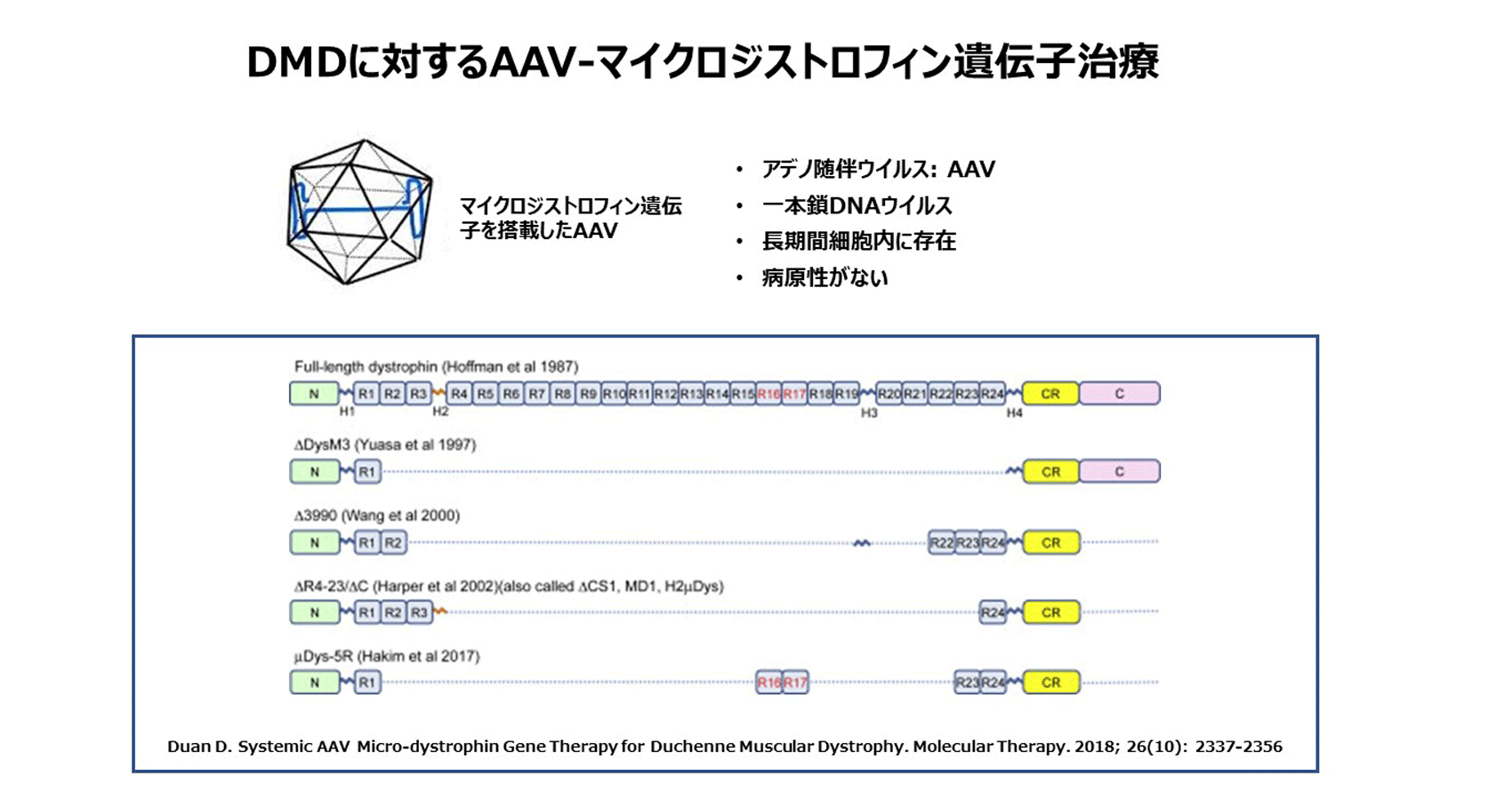
2023年9月現在、ミニ・ジストロフィン/マイクロ・ジストロフィンの遺伝子をAAVベクターで導入する遺伝子治療は、世界で4社が開発中です。そのうち、サレプタ・セラピューティクス(Sarepta Therapeutics, Inc.、以下、サレプタ)、ソリッド・バイオサイエンシズ(Solid Biosciences, Inc.、以下、ソリッドバイオ)、ファイザー(Pfizer, Inc.)の3社では治験が始まっており、サレプタのものは2023年6月に米国で「ELEVIDYS」として、迅速承認されました。それぞれ異なる特徴を持っていますが、いずれも1回の点滴による全身投与で治療します。遺伝子には、「筋肉特異的プロモーター」が備わっているため、全身投与でも短縮型のジストロフィン・タンパク質は筋肉でのみ作られます。ファイザーはミニ・ジストロフィン、サレプタとソリッドバイオはマイクロ・ジストロフィンの遺伝子を搭載しています。また、ソリッドバイオは、5つのロッド・ドメインのうち2つはnNOSの結合部位であるR16とR17のものです。
ベクターに使われているAAVにはいくつかの血清型があり、血清型により感染しやすい(=遺伝子導入が行われやすい)臓器などの特徴が異なります。ソリッドバイオとファイザーで用いられているAAV9という血清型のベクターは、心筋で強い遺伝子発現を誘導するという特徴があります。動物実験では、100%の心筋細胞で導入したマイクロ・ジストロフィンの発現が確認できています。さらにこの発現は、治療の74週後でも、ほとんどの心筋で継続していました。もちろん、骨格筋にも有効で、線維化が起きなかったことも確認されています。DMDでは、心筋細胞のジストロフィン不足により心不全で亡くなられる方が多くいるため、これらの効果は重要だと言えます。迅速承認されたサレプタの「ELEVIDYS」で用いられているAAVベクターの血清型は、AAV-rh74というもので、これは現段階で、最も安全性が高いと言われているAAVベクターです。治験の対象年齢が、ソリッドバイオでは4~17歳、ファイザーでは5~12歳となっていますが、サレプタは生後3か月~7歳と、他の2社に対して低くなっています。治験では、骨格筋でのマイクロ・ジストロフィンの発現増加や筋肉の機能改善などが確認されています。
このように、それぞれに特徴があるわけですが、しかしどれも多かれ少なかれ副作用があります。その中の特に2つが、大きな課題となっています。1つは、遺伝子治療により体内で作られたミニ/マイクロ・ジストロフィンが、体内で異物と認識されて強い炎症が起きるケースがあることです。冒頭の方で、DMDではジストロフィン遺伝子のエクソン45~55に変異が集中していると言いましたが、実は2番目に変異が集中しているのは、エクソン1~エクソン17、18あたりまでです。この部分を欠損している患者さんにとって、N末端部分があるミニ/マイクロ・ジストロフィンは、これまでに経験したことのない新しい分子であるため、異物と捉えられ免疫応答が起きてしまいます。現在、ジストロフィン遺伝子の5’端頭の部分(翻訳されたタンパク質のN末端側)に欠損の変異がある人は、原則として遺伝子治療の対象にしていません。2つ目の課題は、心筋で導入遺伝子が発現しすぎる可能性があることです。心筋でのジストロフィンの発現は重要である一方で、発現が多いと心筋炎などの問題が起こる可能性があります。これらの2つが、DMDの遺伝子治療において改善すべき課題で、今後、発現量のコントロールや、免疫抑制剤の併用による慎重投与などが検討されていくでしょう。
エクソン・スキップ治療と遺伝子治療のほかに、DMDで開発が進んでいる最新治療法があれば教えてください
新たな方法論に基づいた治療法開発の研究がいくつか進んでいます。例えば、ゲノム編集技術を応用し、患者さんの持っている遺伝子そのものを治す治療法の研究が進められています。また、新型コロナウイルスワクチンで用いられた、mRNAワクチンの技術を応用し、ジストロフィンのmRNAを合成して体内に送り込み治療しようという研究も進められています。いずれもまだ研究段階で、臨床試験の段階までには至っていませんが、今後の新たな治療として期待が持てます。
ほかにも、世界でさまざまな治療薬の開発が進んでいますが、注目すべきものの1つとして、Vamorolone(VBP15、バモロロン)が挙げられます。バモロロンは、ステロイド(副腎皮質ホルモン)の代用薬です。ステロイドは、DMDの患者さんに投与すると歩行期間が2年ほど延長するなど、DMDに有効であることが知られています。しかし、ステロイドは肥満、糖尿病、白内障などの副作用があります。そうした副作用をできるだけ抑え、ステロイドの抗炎症作用を示すものがバモロロンで、これは承認が近いと考えています。それから、大鵬薬品が開発中のTAS-205という薬があります。これは、選択的造血器型プロスタグランジンD合成酵素阻害剤といい、筋線維の変性を起こす細胞が集まってくるのを抑制する薬です。宇宙実験により構造が解明されたというユニークな点もあり、日本発の薬として注目されています。バモロロンも、TAS-205も、ジストロフィン遺伝子変異の部位に関わらず、どなたにも投与できる薬です。
最後に武田先生から、遺伝性疾患プラスの読者に一言メッセージをお願い致します
今回は、筋ジストロフィー、特に、DMDに対してどのような治療法が開発されているかをお話ししました。エクソン・スキップ治療や遺伝子治療について、承認されたものも出てきていますが、まだ課題が残っています。しかし、これらの治療を改善する研究は引き続き進められていますし、また、全く新しい発想での治療も開発されてきています。こうした治療が患者さんに届けられる日を待っていてください。皆さんが元気で生活され、活躍されることを心から願っています。
筋肉の機能に必須なタンパク質が、遺伝子変異により正しく作られないことで発症する筋ジストロフィー。中でもDMDは特に重症なタイプで、単一遺伝子疾患でありながら、原因となるジストロフィン遺伝子が巨大であるため、病態も治療法もとても複雑であるとわかりました。しかし、その遺伝子変異の特徴を生かしてさまざまな治療戦略が考案され、核酸医薬品によるエクソン・スキップ治療や遺伝子治療など、実際に国内外で承認薬も出てきている段階だと理解できました。重大な副作用を改善する研究や、ステロイド代用薬の研究なども進められており、今後、治療選択肢が増えていくことが期待できそうです。
今回、武田先生は、国内外の複数の出張で大変お忙しい合間を縫って、取材に協力してくださいました。直近の出張で得られた最新情報まで含めてお話しくださったおかげで、難解な筋ジストロフィーの仕組みや治療について、最新の正しい情報の入った、わかりやすい記事となりました。武田先生、どうもありがとうございました。遺伝性疾患プラスは、今後も筋ジストロフィー治療の最新情報を皆様にお届けしていきます。(遺伝性疾患プラス編集部)





