



どのような病気?
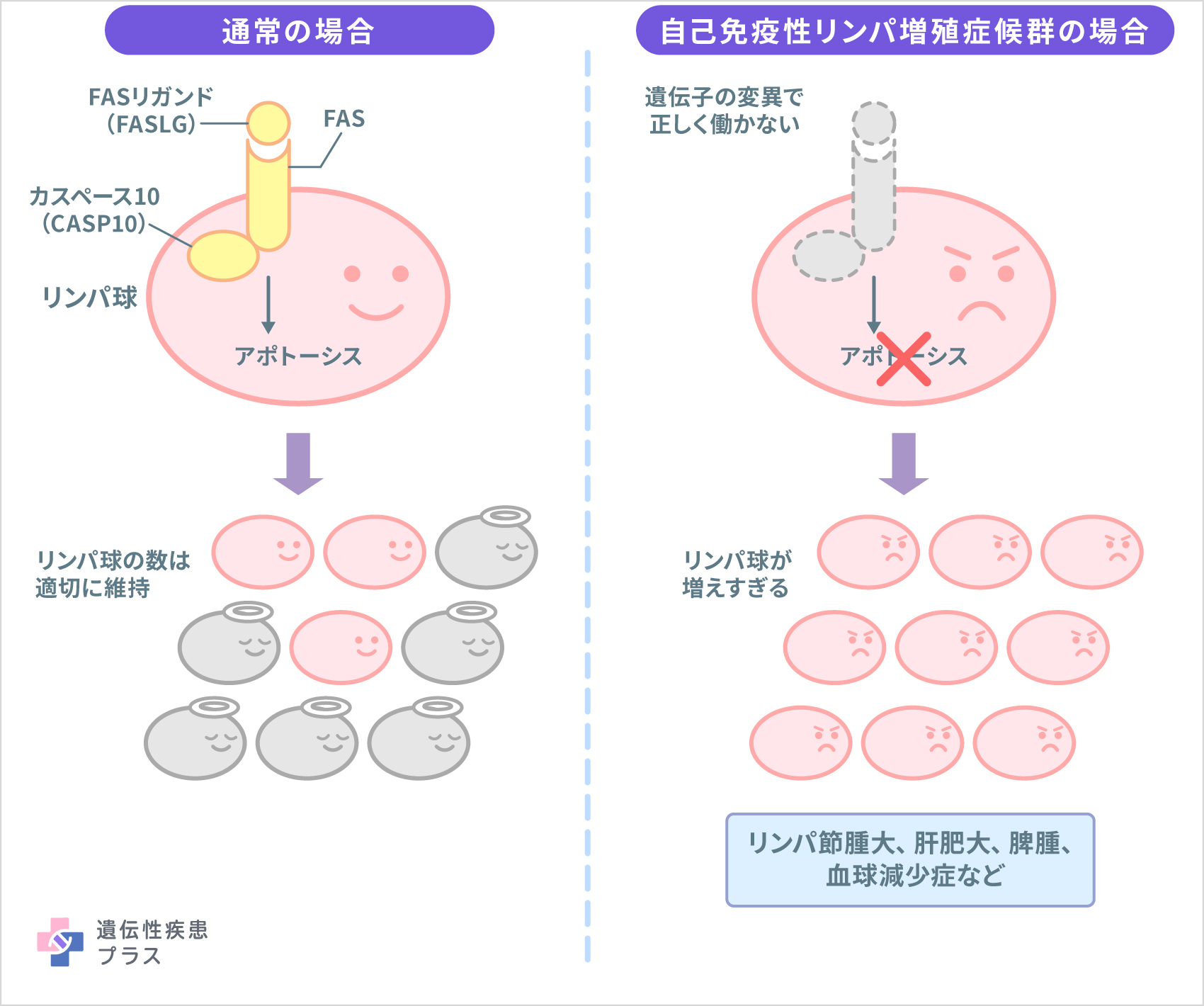
自己免疫性リンパ球増殖症候群(ALPS)は免疫系細胞(リンパ球)の数を適切にコントロールできなくなることによって起こる疾患で、多くの場合遺伝性疾患です。通常、体を守るために増えたリンパ球は役目を終えると、アポトーシスと呼ばれるプログラムされた細胞死によって速やかに除去されます。しかし、ALPSでは、遺伝子の変異が原因でアポトーシスが正常に誘導されず、その結果、リンパ球が過剰に増殖し、リンパ増殖症や自己免疫疾患を発症します。
ALPSにおいて最も特徴的な症状は、持続的なリンパ節腫大、肝腫大、脾腫と、自己抗体や自己反応Tリンパ球増殖による自己免疫疾患の合併です。特に、血液細胞に対する自己抗体が作られることにより、免疫性血小板減少症(ITP、旧病名:自己免疫性血小板減少性紫斑病)、自己免疫性溶血性貧血(AIHA)、自己免疫性好中球減少症(AIN)などの血球減少症がしばしば見られます。頻度は低いですが、腎炎、肝炎、ぶどう膜炎、関節炎など、ほかの臓器にも自己免疫性の炎症を合併することもあります。
どの年齢でも発症する可能性がありますが、多くの患者さんが生後数年で発症するとされています。小児期に発症した自己免疫疾患は成長とともに自然軽快していくものが多いとされますが、成人期までさまざまな自己免疫疾患の合併を認める例もあり、定期的な評価が望ましいとされます。また、ALPSの患者さんは免疫系細胞のがん(リンパ腫)を発症するリスクが高く、他のがんも発症しやすくなる可能性があるため、長期的なモニタリングを十分に行う必要があると考えられます。
患者さんの数は、全世界で400人ほど、国内で20人ほどと推定されています。日本では、原発性免疫不全症候群(指定難病65)に含む形で指定難病対象疾病になっています。また、小児慢性特定疾病にも指定されています。
何の遺伝子が原因となるの?
ALPSの原因として、「FAS」「FASリガンド(FASLG)」「カスペース10(CASP10)」の遺伝子変異が知られています。3つともアポトーシスの誘導に関与するタンパク質の設計図となる遺伝子です。ALPSでは、これら3つの遺伝子のどれかに変異が生じた結果、「Fas誘導性アポトーシス」がリンパ球で正常に働かなくなります。
ALPSは変異が生じた遺伝子によって5つに分類されます。FAS遺伝子変異によるALPSには、生殖細胞系列の変異で発症するもの(親から子へ遺伝するもの)と体細胞レベルの変異で発症するもの(親から子へ遺伝しないもの)が知られ、それぞれ「ALPS-FAS」、「ALPS-sFAS」と分類されています。FASLGの異常によるALPSは「ALPS-FASLG」、CASP10の異常によるものは「ALPS-CASP10」、遺伝子の変異が同定されていないものは「ALPS-U」と分類されます。海外の報告では、患者さんの75%はALPS-FAS、10%がALPS-sFASに分類され、ALPS-FASLGやALPS-CASP10はまれであるとされます。
OMIMや米国国立医学図書館が運営するMedline Plusによると、原因となる遺伝子の変異は、ほとんどが常染色体優性(顕性)遺伝と呼ばれる形式で遺伝するとされます。常染色体優性(顕性)遺伝形式では、両親のどちらかがALPSの場合、子どもは50%の確率で発症します。

しかし、一部の患者さんでは、常染色体劣性(潜性)遺伝形式で遺伝することが報告されています。この場合、それぞれの細胞が2つずつ持つ遺伝子の両方に変異が生じることで発症します。両親は変異のある遺伝子を1つ持つ保因者ではありますが、通常はこの病気を発症しません。保因者である両親から生まれた子どもがALPSである確率は25%です。

また、両親を含めた家族にALPSの病歴がなくても、突然の遺伝子の変化によりALPSを発症することもあります(突然変異による新生変異)。
どのように診断されるの?
ALPS患者さんで特徴的な所見が、末梢血でのTCRαβ鎖発現ダブルネガティブT(DNT)細胞の増加です。持続的なリンパ節腫脹、脾腫または肝腫大、自己免疫疾患などのALPSに特徴的な臨床症状が見られた場合、DNT細胞の評価が行われます。診断基準の必須項目2つと補助項目の一次項目1つ以上を満たした場合にALPSと診断されます。
ALPSの特徴的な臨床症状があるにもかかわらず、DNT細胞の増加を見られない場合には、DNT細胞数を繰り返し測定しつつ、ALPS類縁疾患(ALPSと似た症状を示す別の疾患)を中心とした他疾患の鑑別が行われます。二次項目にある末梢血のIL-10の増加、IL-18の増加、ビタミンB12の増加は、ALPSの補助診断として有用です。また、可溶性FasL(sFasL)の増加はALPS-FASを強く疑う所見です。
ALPSの特徴的な症状やDNT細胞の増加が認められた際には、FAS遺伝子をはじめとするALPS関連遺伝子解析が行われます。ALPSに加えて、ALPS類縁疾患を含めた遺伝子パネル検査が保険適用となっています。また、ALPS-sFASの診断にはDNT細胞だけを集めてFAS遺伝子解析を行う必要があります。
① 必須項目
ⅰ) 6か月以上続く慢性の非悪性・非感染性のリンパ節腫脹または脾腫、もしくはその両方
ⅱ) CD3+TCRαβ+CD4-CD8-T細胞(DNT細胞)の増加(末梢血リンパ球数が正常または増加している場合で、リンパ球全体の1.5%以上またはCD3+T細胞の2.5%以上)
②補助項目
一次項目
ⅰ) リンパ球のFas誘導性アポトーシスの障害
ⅱ) FAS、 FASLG、CASP10のいずれかの遺伝子における体細胞もしくは生殖細胞系列での変異
二次項目
ⅰ) 血漿sFasLの増加(> 200 pg/mL)
ⅱ) 血漿IL-10の増加(> 20 pg/mL)
ⅲ) 血清または血漿ビタミンB12の増加(> 1500 pg/mL)
ⅳ) 典型的な免疫組織学的所見(傍皮質T細胞過形成)
ⅴ) 自己免疫性血球減少(溶血性貧血、血小板減少または好中球減少)
ⅵ) 多クローン性IgG増加
ⅶ) 自己免疫の有無に関わらず非悪性/非感染性のリンパ増殖症の家族歴がある
特徴的な症状やDNT細胞の増加を認めるにもかかわらず、ALPS関連遺伝子に変異が認められない症例も存在します。そのような症例では、リンパ球のFas誘導性アポトーシスの障害が確認されます。Fas誘導性アポトーシスの評価が可能な施設は限られますが、他の疾患と鑑別する上で有用な検査です。
どのような治療が行われるの?
リンパ球増殖の制御と自己免疫性血球減少症に対する治療が中心になります。血球減少症に対しては、副腎皮質ステロイド投与が有効とされており、免疫グロブリン大量療法が併用される場合もあります。ステロイド治療が長期化する場合は、免疫抑制剤の併用が必要になります。海外では、ミコフェノール酸モフェチル(MMF)、リツキシマブ、mTOR阻害薬などによる治療が試みられており、特にmTOR阻害薬の高い有効性が報告されています。国内では、リツキシマブが慢性ITPに対して保険適用となっていますが、自己免疫性血球減少症に対するMMF、mTOR阻害薬の使用は保険適用外です。
脾臓の機能亢進がコントロールできない場合や脾破裂のリスクを伴う場合には、脾臓摘出(脾摘)が行われる場合もあります。ただし、低年齢時に脾摘が行われた症例で、致死的な敗血症を合併した例が報告されており、脾摘については慎重に検討する必要があるとされています。根治療法として造血幹細胞移植が考えられますが、基本的に生命予後は良好な疾患であり、加齢とともに症状が軽快することが多いとされることから、限定的な治療選択となります。
どこで検査や治療が受けられるの?
日本で自己免疫性リンパ増殖症候群の診療を行っていることを公開している、主な施設は以下です。
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
自己免疫性リンパ増殖症候群(原発性免疫不全症候群)の患者会で、ホームページを公開しているところは、以下です。
参考サイト
- 難病情報センター
- 小児慢性特定疾病情報センター 自己免疫性リンパ増殖症候群
- 自己免疫性リンパ増殖症候群 (ALPS) の診療ガイドライン作成について
- 自己免疫性リンパ増殖症候群 (ALPS) の診療ガイドライン改訂について
- MedlinePlus
- Genetic and Rare Diseases Information Center
- Online Mendelian Inheritance in Man(R) (OMIM(R))
- Orphanet






