どのような病気?
フェニルケトン尿症(PKU)は、血液中のフェニルアラニンと呼ばれる物質の量が増加する遺伝性疾患で、先天代謝異常症と呼ばれる疾患の1つです。高フェニルアラニン血症(HPA)とも呼ばれます。
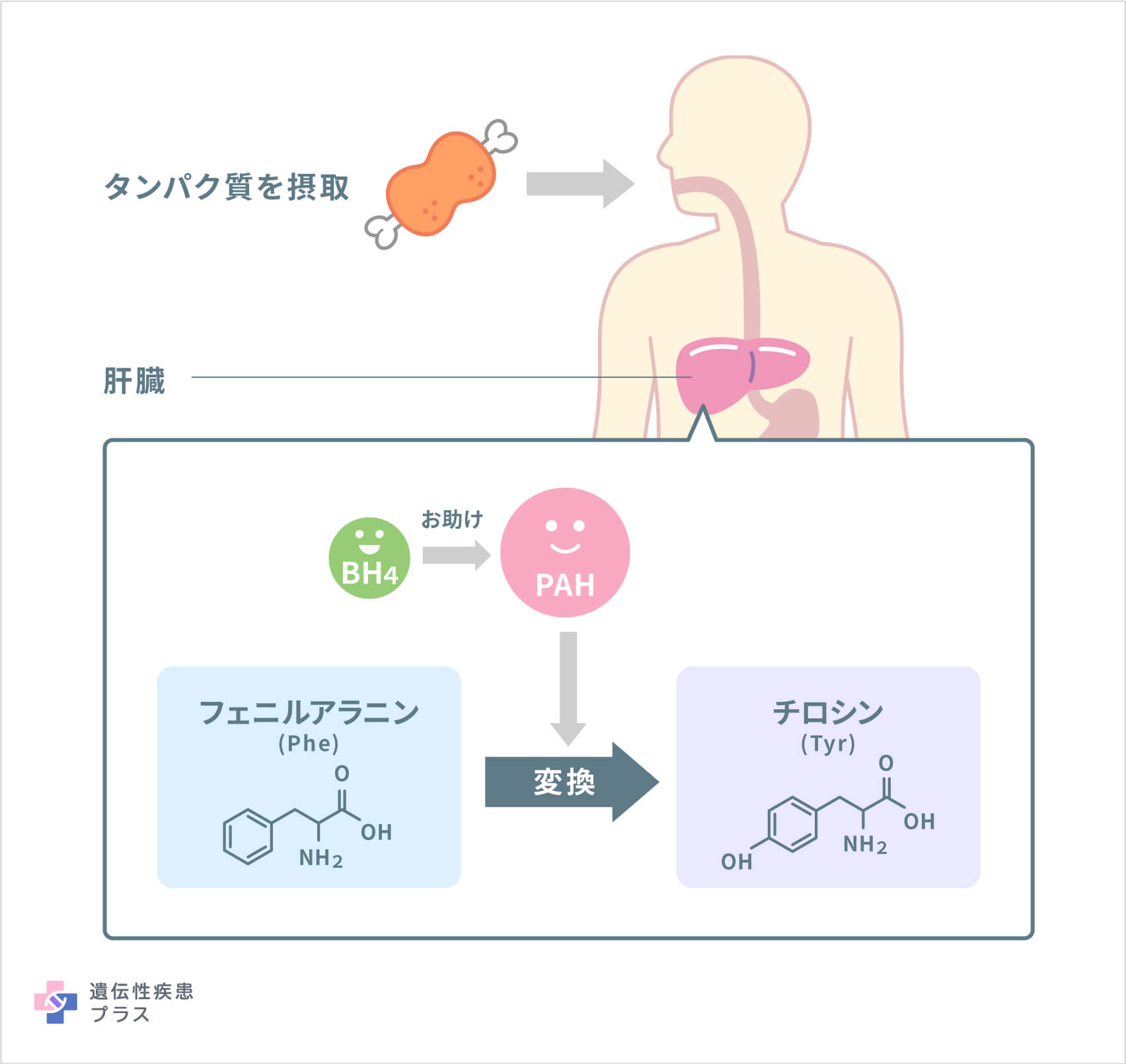
フェニルアラニンは、タンパク質の構成要素であるアミノ酸の一種で、さまざまなタンパク質と、いくつかの人工甘味料に含まれています。このフェニルアラニンは、アミノ酸の中でも、ヒトの体内で合成できず食物から摂取する必要がある「必須アミノ酸」の1つです。しかし、ヒトは食べて取り込んだフェニルアラニンを全ては必要としないので、体は余分なフェニルアラニンを別のアミノ酸であるチロシンに変換します。PKUは、肝臓でフェニルアラニンをチロシンに変える酵素である「フェニルアラニン水酸化酵素(PAH)」の働きが生まれつき弱く、体内にフェニルアラニンが有害なレベルまで蓄積し、チロシンが少なくなる病気です。体内で増えたフェニルアラニンは、フェニルケトン体として尿に排泄されます。この尿は特有の尿臭があり、ネズミ尿臭、カビ臭、といった表現を使われる場合があります。
PKUの重症度はさまざまで、診断時の血漿中フェニルアラニン値により、高い方から「古典的PKU(20mg/dL以上)」「軽症PKU(10mg/dL以上20mg/dL未満)」「軽症高HPA(10mg/dL未満)」に分類されます。酵素(PAH)の働きは、補酵素「テトラヒドロビオプテリン(BH4)」に助けられています。PAHに異常があっても、BH4に反応して血中のフェニルアラニン値が低下する病型があり、これは「BH4反応性PKU(HPA)」と呼ばれています。古典的PKUでは、治療を受けずにフェニルアラニンが体に蓄積し、それが持続すると、脳の発達障害をきたします。また、チロシンが少なくなると、色素が正常に作れなくなり、髪の毛や皮膚の色が薄くなります(赤毛、色白など)。
PKUは、生まれて間もなくはほとんど症状がありませんが、治療を受けずに高い血中フェニルアラニン値が持続すると、通常生後数か月から2歳頃までに発達遅滞などの神経症状を発症します。小頭症、てんかん、重度の精神発達遅滞、行動上の問題などが見られるほか、画像検査によって脳萎縮や白質病変が認められることがあります。
また、まれにBH4が正しく作れなくなり欠乏する異常により、血中のフェニルアラニン値が高くなる人もいます(BH4欠損症)。BH4欠損症では、さらに神経の働きを伝える物質(ドーパミン、ノルアドレナリン、アドレナリン)も少なくなるため、より重い精神発達の障害が早期から見られます。
| フェニルケトン尿症で見られる症状 |
|---|
99~80%で見られる症状 尿中アミノ酸レベル高値 |
79~30%で見られる症状 早期・重度の精神遅滞 |
29~5%で見られる症状 心臓血管系の形態異常 |
割合は示されていないが見られる症状 攻撃的な行動、不安、注意欠如・多動症(ADHD)、青い目、白内障、脳の石灰化、うつ病、乾燥肌、金髪、色白な肌、高フェニルアラニン血症、反射亢進、尿中の馬尿酸値の上昇、易怒性、小頭症、強迫性行動、フェニルピルビン酸血症、精神病、フェニルアラニンヒドロキシラーゼ値低下、強皮症、てんかん発作、自傷行為 |
PKUの女性が妊娠し、低フェニルアラニン食などでフェニルアラニンの値を制御していなかった場合、流産のリスクが高くなります。また、出生前に高値のフェニルアラニンにさらされた赤ちゃんは、知的障害のリスクが高くなります。また、生まれた赤ちゃんは、出生体重が低かったり、他の子どもより成長が遅かったりする場合があります。
PKUは、新生児マススクリーニング検査の対象疾患として、厚生労働省が各自治体に勧奨している20の疾患のうちの1つです。PKUの患者さんは、日本で毎年20人前後見つかっており、発生頻度は約7~8万出生に1人とされています。こども家庭庁成育局母子保健課の先天性代謝異常等検査実施状況(令和4年度)によれば、PKUの昭和52~令和4年度までの発見患者数の合計は815人と報告されています。
BH4欠損症は、約170万出生に1人とされており、患者さんは日本に50人程度いると推定されています。
PKUは、国の指定難病対象疾患(指定難病240)、および、小児慢性特定疾病の対象疾患です。BH4欠損症は、PKUに含む形で指定難病対象疾患となっています。また、「ビオプテリン代謝異常症」として小児慢性特定疾病の対象疾患となっています。
何の遺伝子が原因となるの?
PKUの原因遺伝子として12番染色体の12q23.2という位置に存在する「PAH遺伝子」が見つかっています。PAH遺伝子は、フェニルアラニンヒドロキシラーゼ(PAH)と呼ばれる酵素の設計図となる遺伝子です。PAHは、補酵素BH4の助けを借りつつ、アミノ酸であるフェニルアラニンを別のアミノ酸であるチロシンに変換する役割を果たします。遺伝子変異により、PAHの活性が低下すると、食事から摂取したフェニルアラニンは正しく処理されず、血液やその他の組織に有害なレベルまで蓄積する可能性があります。こうした仕組みで、PKUの症状が現れます。最も重症な古典的PKUでは、PAHの活性が大幅に低下するか、PAH自体が存在しないような状況になっています。酵素の活性がそれよりも保持されている場合、より軽症なPKUとなります。
BH4欠損症では、 BH4の合成や再生に関わる、GCH1、PCBD1、PTS、QDPRなどのいくつかの遺伝子の変異が原因になると考えられています。
PKUは、常染色体性劣性(潜性)遺伝形式で親から子へ遺伝します。人間が2本1セットで持っているPAH遺伝子のうち、両親がともに1本ずつ変異を有していた場合、子どもは4分の1の確率で2本とも変異を有してPKUになります。また、2分の1の確率で1本変異を有し発症はしない「保因者」となり、4分の1の確率でPKUを発症せず保因者でもなく(変異した遺伝子を持たず)生まれます。

どのように診断されるの?
PKUは、新生児マススクリーニング検査で高フェニルアラニン血症として発見されます。具体的には、アミノ酸分析(HPLC法)により、血中フェニルアラニン値を測定し、2mg/dL(120µmol/L)以上(基準値0.7~1.8mg/dL)となった場合、「生化学診断例」となります。
その後、BH4欠損症との鑑別診断が行われます。BH4欠損症との鑑別は、血液・尿のプテリジン分析(BH4欠損症で異常パターンが見られる)と、乾燥濾紙血のジヒドロプテリジン還元酵素:DHPR活性の測定(BH4欠損症に含まれるDHPR欠損症ではDHPR活性の著しい活性低下を認める)により行われます。必要に応じてBH4経口負荷試験やPAH遺伝子の解析も行われます。
これらの検査によって、各種PKUやBH4欠損症と病型分類できた場合、確定診断となります。
どのような治療が行われるの?
PKUは、低フェニルアラニン食事療法を継続し、血液中のフェニルアラニンを一定の範囲にコントロールすることで、発症を予防することができます。食事療法では、タンパク質を制限してフェニルアラニンの摂取を抑え、不足する他のアミノ酸を治療粉乳等で補います。BH4反応性PKUはBH4の補充療法が行われます。生まれてすぐの検査で血中フェニルアラニン値が高いとわかったら、早期に食事療法が開始され、数日のうちに血中フェニルアラニン値を10mg/dL以下までに下げます。その後、血中フェニルアラニン値が2~4mg/dLまで低下するように、フェニルアラニン摂取量が調節されます。こうした初期の治療は、通常入院で行われます。BH4欠損症と診断された場合、食事治療に加え、BH4と神経伝達物質の補充療法を受けます。
これらの治療は生涯にわたって続ける必要がありますが、治療を正しく継続すれば、正常に発育・発達します。ですので、食事療法を注意深く続けていくことが重要です。BH4欠損症では薬の飲み忘れが致命的な症状の発症につながります。お子さんが自分で薬を飲むようになったら、毎日きちんと飲むように教えることも重要です。
血中フェニルアラニン値の維持範囲は、乳児期前半までは2~4mg/dL、乳児期後半~小学生前半は2~6mg/dL、小学生後半は2~8mg/dL、中学生以降は2~10mg/dLとされています。
女性では、妊娠する前から出産するまでの間、赤ちゃんに影響しない程度の血中フェニルアラニン値(5mg/dL程度)を維持することが必要です。妊娠を希望する場合は普段より厳しい食事治療が必要になるので、日頃から定期的に通院して、血中フェニルアラニン値をしっかり管理していきましょう。
BH4反応性PKUの人が妊娠する場合、BH4療法によって赤ちゃんへの高フェニルアラニン値の影響が防げると考えられますが、BH4療法が赤ちゃんに対して安全かどうかは、まだ確実な結論は出ていません。
PAHの酵素活性を代替する治療薬として、2023年5月より「パリンジック(R)皮下注 2.5mg/10mg/20mg」(一般名:ペグバリアーゼ(遺伝子組換え))が成人のフェニルケトン尿症治療薬として発売され使用可能となりました。同剤は、既存治療を行っても血中フェニルアラニン濃度のコントロールが不十分な場合に使用が考慮されます。
どこで検査や治療が受けられるの?
日本でフェニルケトン尿症の診療を行っていることを公開している、主な施設は以下です。
- 北海道医療センター
- 秋田大学大学院医学系研究科医学専攻機能展開医学系 小児科学講座
- 東北大学病院
- 千葉県こども病院 代謝科
- 国立成育医療研究センター
- 藤田医科大学医学部 小児科学代謝外来
- 大阪公立大学医学部附属病院 小児科・新生児科
- 島根大学医学部附属病院 臨床遺伝診療部
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
フェニルケトン尿症の患者会で、ホームページを公開しているところは、以下です。
LINEオープンチャットを活用した「QLifeフェニルケトン尿症オンラインコミュニティ」では、フェニルケトン尿症の当事者・ご家族が参加し、ざっくばらんに情報交換をしています。よろしければご参加ください。
参考サイト
- 難病情報センター
- 小児慢性特定疾病情報センター フェニルケトン尿症(高フェニルアラニン血症)
- 小児慢性特定疾病情報センター ビオプテリン代謝異常症
- MedlinePlus
- Genetic and Rare Diseases Information Center
- こども家庭庁成育局母子保健課、先天性代謝異常等検査実施状況(令和4年度)、特殊ミルク情報 59号(2024年 2月)