



どのような病気?
脊髄性筋萎縮症(SMA)は、脊髄の運動神経細胞(脊髄前角細胞という運動ニューロン)に障害が起こり、体幹や手足の筋力低下と筋委縮が起こる、進行性の遺伝性疾患です。国の指定難病対象疾病になっています(指定難病3)。国際SMA協会により、発症年齢と臨床経過に基づいて、4つの病型に分類されています。
I型は、6か月未満の乳児期に発症する重症型で、別名「ウェルドニッヒ・ホフマン(Werdnig-Hoffmann)病」です。最高到達運動機能は臥床までで、自力でお座り(座位)をすることができません。ミルクを飲むことが難しく(吸いが弱く飲みが悪い)、誤嚥や呼吸不全を伴います。舌の細かいふるえも見られます。多くの場合、気管内挿管や気管切開と人工呼吸管理を行います。力が入らずだらりとしている、「フロッピーインファント」という状態であることが特徴として挙げられます。
II型は、1歳半(18か月)未満の乳児~幼児期に発症する中間型で、別名「デュボビッツ(Dubowitz)病」です。最高到達運動機能は、お座りまでで、支えなく立ったり歩いたりすることはできません。舌や手指のふるえが見られ、成長とともに関節の拘縮や背骨の曲がり(側弯)が目立ってきます。呼吸器の感染症を起こして肺炎や無気肺(肺に空気が入らない状態)になると、呼吸不全になることがあります。
III型は、1歳半から20歳までの小児期~若年期に発症する軽症型で、別名「クーゲルベルグ・ウェランダー(Kugelberg-Welander)病」です。最高到達運動機能は、立って歩くところまでいくのですが、次第に転びやすい、歩けない、立てないなどの症状が出てきて、手を上げるのも困難になるケースがあります。小児期以前に発症した人では、側弯が見られます。
IV型は、20歳を過ぎ、成人になってから発生するもので、それまでの運動機能は正常です。側弯は見られず、発症年齢が遅いほど、進行のスピードも遅くなります。
乳児期から小児期にSMAが発症する人は、10万人に1~2で、I型は出生2万人に1人程度とされています。病気のなりやすさに男女差はありません。
何の遺伝子が原因となるの?
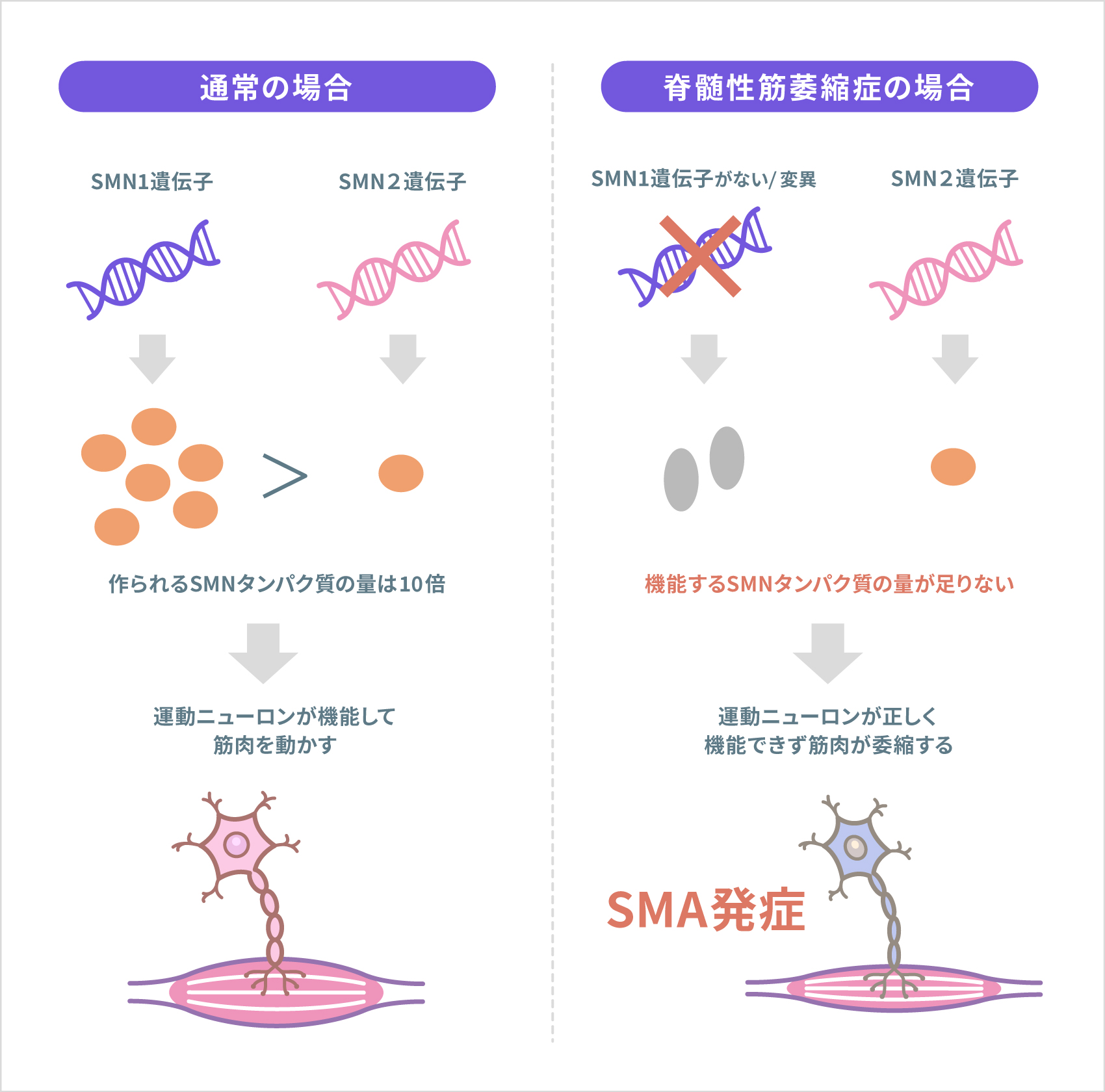
SMAの原因として、「SMN1遺伝子」の変異(欠失を含む)が見つかっています。SMN1遺伝子から作られるタンパク質は、「survival motor neuron(SMN)タンパク質」といい、運動ニューロンの生存維持に重要な役割を担っています。このタンパク質は、体全体に存在しますが、脊髄に特に多く存在します。SMN1遺伝子に変異があり、正常なSMNタンパク質が作れないと、SMAが発症します。I型とII型の95%、III型の約半数、IV型の10~20%にSMN1遺伝子の変異が認められています。
人間は、1番から22番までの常染色体と性染色体を有していますが、SMN1遺伝子は、第5染色体の5q13という位置に存在するとわかっています。同じ5q13という位置には、SMN2遺伝子という、SMN1遺伝子によく似た遺伝子が存在しますが、SMN2遺伝子からは、正常なSMNタンパク質が、SMN1遺伝子の10分の1しか作られないため、これだけで機能を補うことはできません。
早期に重症な呼吸障害を示すI型の一部では、IGHMBP2遺伝子という、細胞分裂に関わる遺伝子の変異が見つかる場合もあります。また、III、IV型においては、SMN1遺伝子変異が見つからない場合も多く、他の原因も考えられています。
SMN1遺伝子の変異は、常染色体劣性(潜性)遺伝形式で、親から子へ遺伝します。人間が2本1セットで持っているSMN1遺伝子のうち、両親がともに1本ずつ変異を有していた場合、子どもは4分の1の確率で2本とも変異を有してSMAを発症します。また、2分の1の確率で1本変異を有し発症はしない「保因者」となり、4分の1の確率でSMAを発症せず保因者でもなく(変異した遺伝子を持たず)生まれます。なお、IV型は、親には遺伝子変異がなく、子どもで初めて発症する「孤発例」が多いとされています。

どのように診断されるの?
SMAは、医師がSMAと診断するための「診断基準」があります。したがって、病院へ行き、必要な問診や検査を受けた後、主治医の先生がそれらの結果を診断基準に照らし合わせ、結果的にSMAかそうでないかの診断をすることになります。重症度分類もされます。具体的には、特徴的な臨床症状と、臨床検査の所見(血清CK値検査や、筋電図などの電気生理学的検査で運動神経の働きを調べるなど)を踏まえたうえで、その他の似たような症状を呈する病気ではない場合にSMAと診断されます。遺伝子検査で、SMN1遺伝子、IGHMBP2遺伝子など、SMAで見られる遺伝子異常を調べる場合もあります。SMAに似ているため、鑑別が必要な遺伝性疾患は、筋萎縮性側索硬化症、球脊髄性筋萎縮症、脳・脊髄腫瘍、脊髄疾患、頸椎症、椎間板ヘルニア、脊髄空洞症など、末梢神経疾患、多発性神経炎(遺伝性、非遺伝性)、多巣性運動ニューロパチーなど、筋疾患(筋ジストロフィー、多発筋炎など)、感染症に関連した下位運動ニューロン障害(ポリオ後症候群など)、傍腫瘍症候群、先天性多発性関節拘縮症、神経筋接合部疾患などです。
SMAは進行性の病気です。早期診断のために、新生児マススクリーニング(赤ちゃんの先天性代謝異常等の病気をみつけるための検査)にSMA検査を追加する動きが日本でも既に始まっています。
どのような治療が行われるの?
SMAは、日本を含めた全世界で、治療薬の開発研究や治験(臨床試験)が行われています。その成果の1つとして、2017年には核酸医薬「ヌシネルセン」(製品名:スピンラザ)が、SMA治療薬として国内で承認され、販売されています。この薬は、「アンチセンスオリゴヌクレオチド」と呼ばれる種類の核酸医薬で、遺伝子からタンパク質が作られる過程にはたらくことで、SMN2遺伝子から作られる正常なSMNタンパク質の量を10%より増やし、機能を補う狙いの薬です。定期的に髄腔内に注射して治療します。
また、2020年3月に、遺伝子治療薬「オナセムノゲン アベパルボベク」(製品名:ゾルゲンスマ)が国内で承認され、同年5月に保険適用となり発売されました。この薬は、「ベクター」と呼ばれる、遺伝子を細胞内に運び込む手段を用いて、運動ニューロンなどに正常なSMN1遺伝子を人工的に導入し、SMNタンパク質が十分な量、作られるようにする薬です。治療は、たった1回の点滴で完了します。SMAは進行性の病気ですが、この薬の効果は、早期に治療を行うほど高いことがわかっています。
さらに、2021年8月には、脊髄性筋萎縮症治療剤「リスジプラム」(製品名:エブリスディ(R)ドライシロップ60mg)が、脊髄性筋萎縮症を効能又は効果として、国内で販売開始されました。この薬は、SMN2遺伝子からSMNタンパク質が多く作られるようにデザインされた、「SMN2スプライシング修飾剤」と呼ばれる経口薬です。通常、1日1回、食後に飲んで治療します。
症状に対する治療も行われます。筋力に合わせて運動訓練や、関節拘縮予防のためのリハビリテーションが行われます。III型では、歩ける状態をできるだけ長く維持するためにもリハビリテーションは重要です。必要に応じて、装具の使用なども検討されます。自力での移動が困難な場合には、手の力も弱いため、電動車いすを利用します。例えば、II型では3歳頃には電動車いすを動かして、家や外で友達や家族と一緒に過ごすことができるようになります。こうしたリハビリテーションは、小児神経医、神経内科医、整形外科医、理学療法士などが連携して取り組みます。
I型やII型の乳幼児の場合、咳の力が弱い、呼吸器感染を繰り返す、哺乳や食事の後に疲れた様子を見せる、体重が増えないなどの徴候が見られないかどうか、常に注意を払う必要があります。一方で、食欲の良好なII型やIII型の幼児では、肥満にならない注意も必要です。
I型やII型で授乳が困難な場合には、経管栄養や胃ろうを行う場合があります。呼吸器感染や無気肺を繰り返す人が多いのですが、急速に進行したり、人工呼吸管理が必要になったりする場合があるので、注意が必要です。鼻マスク人工換気法(NIPPV)は、乳児での使用は難しいですが、有効と考えられています。呼吸器感染を起こした場合には、カフマシン(排痰補助装置)の使用や、肺の理学的療法による排痰ドレナージ(医師の指示に基づいて、体位などを工夫して痰を出す手技)が有効とされています。
また、脊髄性筋萎縮症(SMA)の診断を受けられた方々が、治験などにご参加いただけるための患者登録システム「SMARTコンソーシアム」が、厚生労働省の研究班などにより設立されています。SMARTコンソーシアムは、SMAの根本治療達成という目標に向け、治療法のさらなる発展・充実とケアの拡充を目指す目的で運営されています。
どこで検査や治療が受けられるの?
日本で脊髄性筋萎縮症の診療を行っていることを公開している、主な施設は以下です。
- 東京女子医科大学遺伝子医療センターゲノム診療科
- 東京慈恵会医科大学附属病院小児神経外来
- NCNP病院 国立精神・神経医療研究センター
- 新潟大学脳研究所 脳神経内科
- 新潟病院
- 愛知医科大学医学部小児科
- 京都府立医科大学附属病院神経外来(小児科)
- 関西医科大学 小児科学教室
- 久留米大学病院
- 福岡大学病院遺伝医療室
- 鹿児島大学大学院医歯学総合研究科 神経病学講座 脳神経内科・老年病学
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
脊髄性筋萎縮症の患者会で、ホームページを公開しているところは、以下です。
参考サイト
・参考文献:医学書院 医学大辞典 第2版








