



どのような病気?
筋萎縮性側索硬化症(ALS)は、筋肉を動かし、かつ運動をつかさどる神経の「運動ニューロン」が、選択的に変性・消失していく病気です。運動ニューロンが障害を受けると、脳からの「手足を動かしなさい」という命令が伝わらなくなります。その結果、手足・のど・舌の筋肉や、呼吸に必要な筋肉がだんだん痩せて力がなくなっていきます。多くの場合、最初、手指が使いにくくなり、握力の低下やつまずきやすさを自覚し、ひじから先の力が弱くなって、筋肉が痩せてきます。人によっては、話しにくかったり、食べ物が飲み込みにくかったりという症状(球麻痺、きゅうまひ)から始まります。やがて、全身の筋肉が痩せて力が入らなくなり、歩けなくなります。のどの筋肉が弱くなると、声が出しづらくなったり、飲み込みづらくなったりします(嚥下障害)。呼吸の筋肉も次第に弱くなり、十分に呼吸ができなくなってきます。一方、目の動き、体の感覚、視力や聴力、内臓機能などは、一般的に問題なく保たれます。
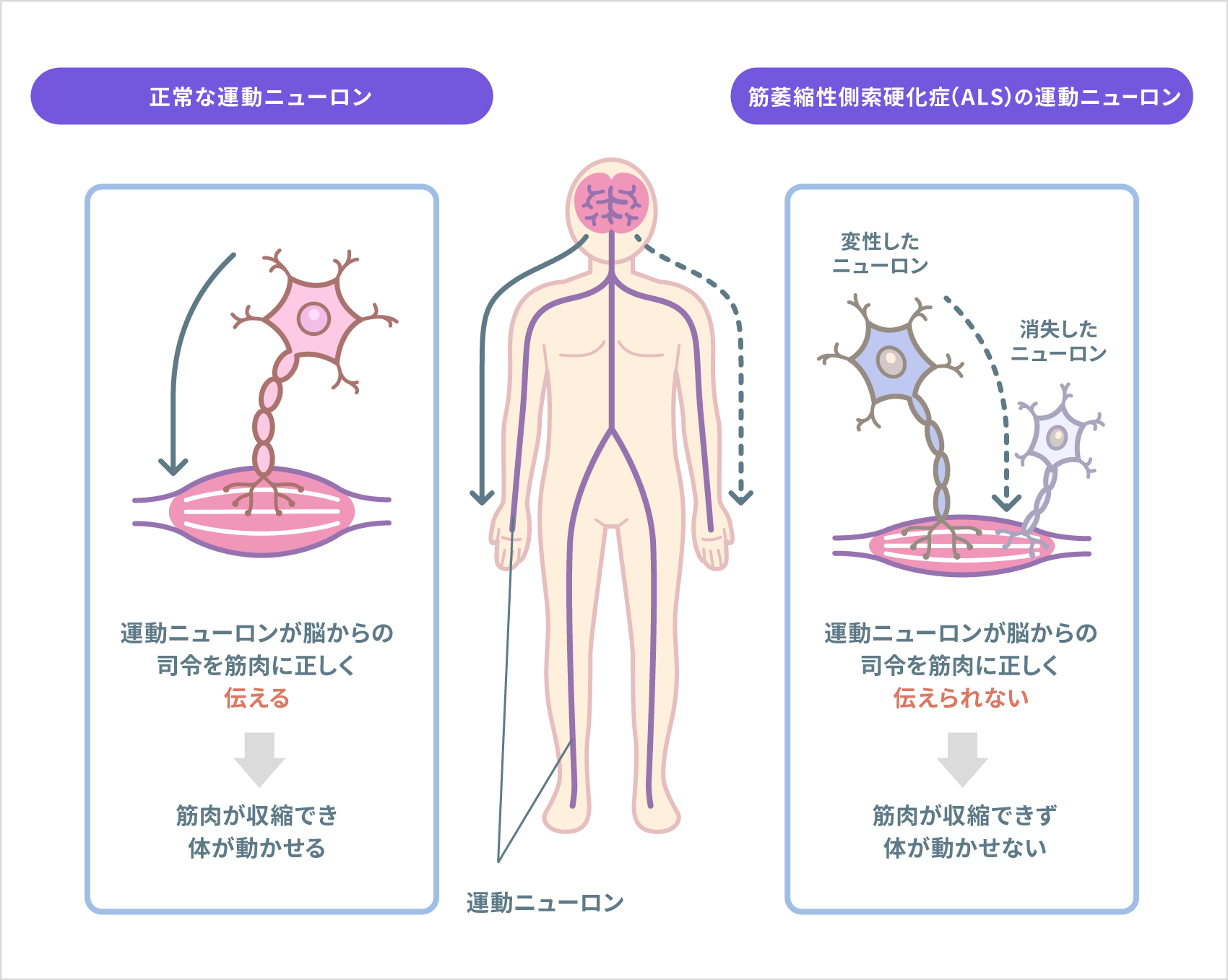
ALSは進行性の病気で、症状の進行は速いとされているものの、正確な調査はなく、個人差は大きいと知られています。発症原因は不明ですが、フリーラジカル(活性酸素など)が細胞を傷つけることや、過剰なグルタミン酸が神経に毒性を及ぼすこと(グルタミン酸毒性)などが、可能性として挙げられています。人工呼吸器を使わない場合、発症から2~5年で多くの患者さんが亡くなりますが、人工呼吸器を使わなくても10数年かけてゆっくり進行する人もいます。反対に、もっと進行が速い人もいます。
多くのALSで、発病の原因は今のところ不明ですが、遺伝子異常等との関連が考えられています。全体の約5%は、「家族性ALS」と呼ばれ、この中には、原因となる遺伝子異常が判明しているものもあります。家族性ALSは、親から子へ遺伝しますが、それ以外の多くは、遺伝しないALS(孤発性ALS)です。孤発性ALSは、環境要因と、遺伝的要因(体質的なALSのなりやすさなど)が、複数、複雑に絡んで発症すると考えられており、研究は進められていますが、まだ解明されていません。
ALSの発症年齢は、10歳代後半から80歳代におよびますが、一般には中年以降で、最も発症しやすいのは60~70歳代と知られています。ALS患者さんの数は、男性が女性の1.2~1.3倍で、男性でやや多くなっています。また、家族性か孤発性かに関わらず、ALS患者さんの約5%で、認知症(前頭側頭型認知症)が見られます。
ALSは、筋肉に症状があることから、筋ジストロフィーと混同されがちな部分もありますが、両者は原因遺伝子や発症の仕組みなどが異なる、別の病気です。
筋委縮性側索硬化症(ALS)は、指定難病対象疾患(指定難病2)です。
何の遺伝子が原因となるの?
家族性ALSの約2割では「スーパーオキシド・ジスムターゼ(SOD1)」という酵素の遺伝子に異常が見つかっています。SOD1は、過剰に存在すると細胞を障害してしまう「活性酸素」を処理する酵素です。
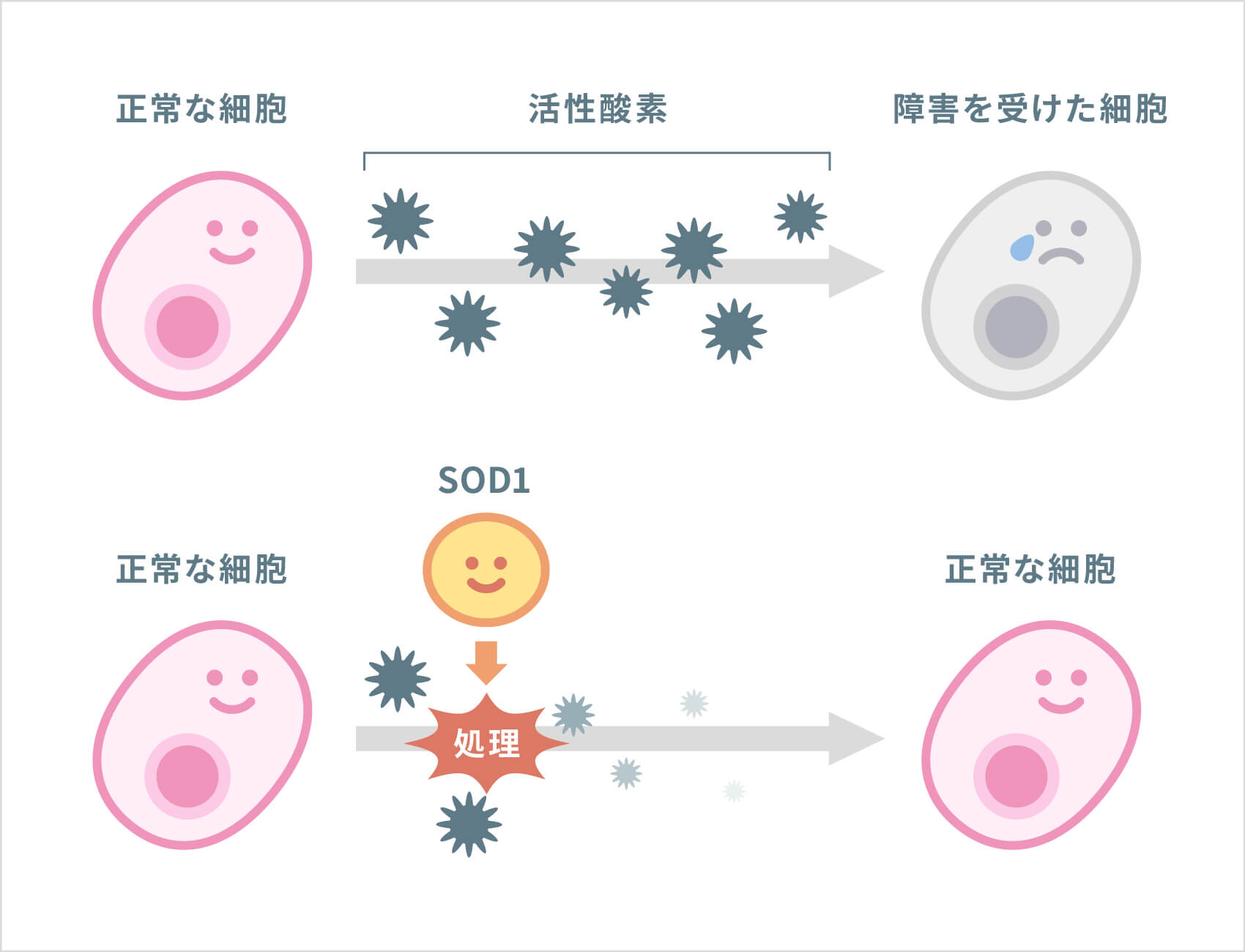
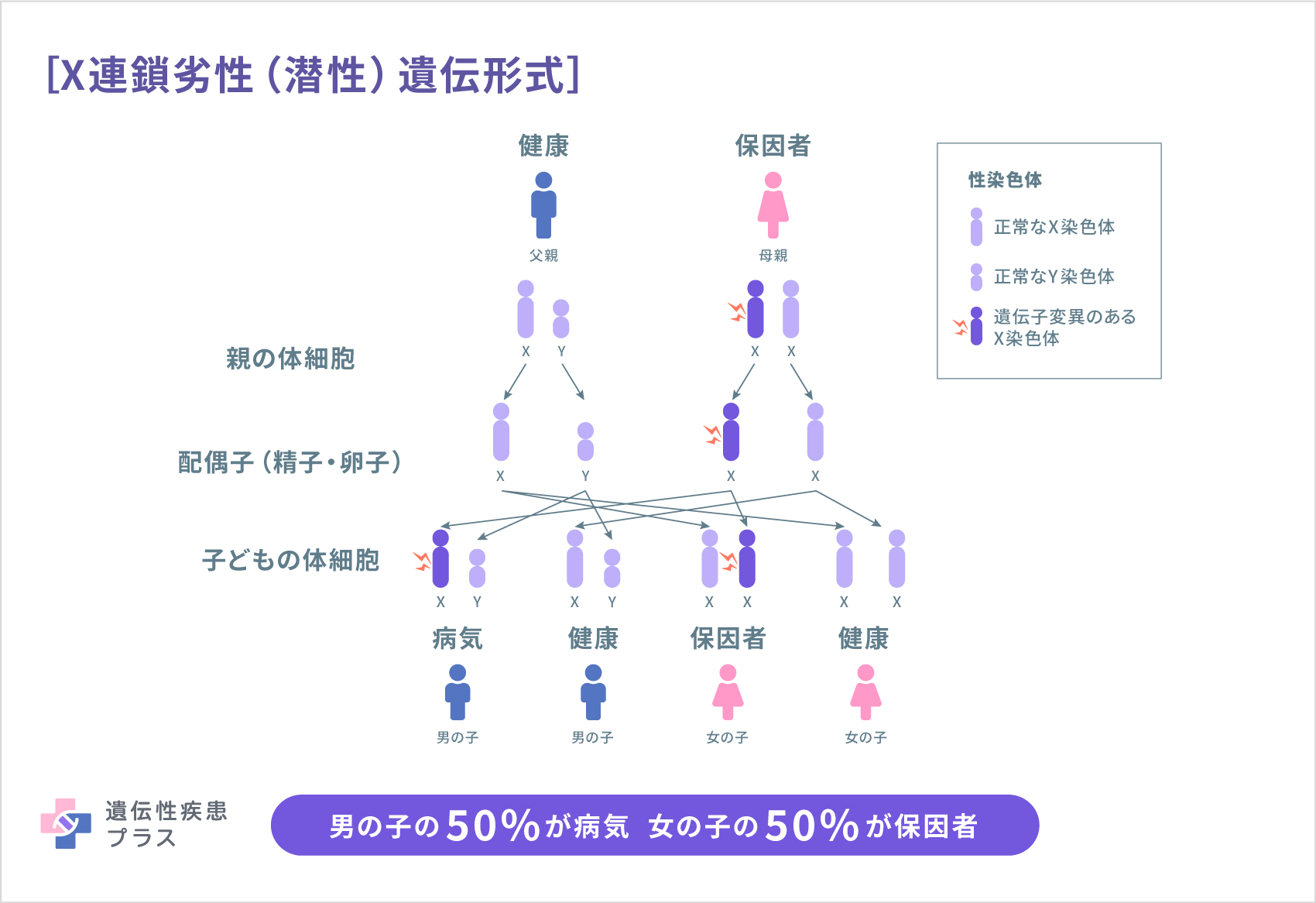
異常なSOD1遺伝子から作られたSODは、運動神経の細胞死を引き起こすことが、これまでの研究でわかってきています。SOD1遺伝子の異常は、常染色体優性(顕性)遺伝です。その他にも、常染色体劣性(潜性)遺伝する原因遺伝子、X連鎖型遺伝する原因遺伝子など、近年、次々とALSの原因遺伝子が報告されてきています(下表参照)。
これまでに見つかっている、家族性ALSの原因遺伝子の例
| 家族性ALS患者の割合(%) | 遺伝子 | 遺伝形式 | 遺伝子から作られるタンパク質の機能 |
|---|---|---|---|
| 20 | SOD1 | 常染色体優性(顕性) | スーパーオキシド・ジスムターゼ:銅と亜鉛の分子を利用して活性酸素を処理する |
| ~4 | FUS | 常染色体優性(顕性) | RNA結合タンパク質FUS:mRNAの長さ調節などにより細胞のさまざまな機能に関わる |
| まれ | ANG | 常染色体優性(顕性) | アンジオゲニン:血管を新生する |
| まれ | FIG4 | 常染色体優性(顕性) | ポリホスホイノシチドホスファターゼ:脱リン酸化酵素の1つで、細胞のさまざまな機能に関わる |
| まれ | ALS2 | 常染色体優性(顕性) | アルシン:GTPaseと呼ばれる、さまざまな細胞の機能に関わるタンパク質を活性化するタンパク質で、運動ニューロンに豊富に含まれる |
| まれ | OPTN | 常染色体優性(顕性) | オプチニューリン:他のタンパク質と相互作用して、細胞の形態形成など、複数の機能に関わっている |
| まれ | UBQLN2 | X連鎖性 | ユビキリン2:細胞内の不要なタンパク質を分解する機能に関わっている |
孤発性ALSに対しても今、なりやすさに関連する遺伝子(疾患感受性遺伝子)を探すため、大勢の患者さんを対象としたゲノム解析の研究が進行中です。


どのように診断されるの?
ALSには、医師がALSと診断するための「診断基準」があります。したがって、病院へ行き、必要な問診や検査を受けた後、主治医の先生がそれらの結果を診断基準に照らし合わせ、結果的にALSかそうでないかの診断をすることになります。具体的には、特徴的な臨床症状(触診によるバビンスキー反射などを含む)と、筋電図などの電気生理学的検査で運動神経の働きなどを調べたうえで、その他の似たような症状を呈する病気ではない場合にALSと診断されます。遺伝子検査で、SOD1遺伝子など、家族性ALSで見られる遺伝子異常を調べる場合もあります。ALSに似ているため、鑑別が必要な遺伝性疾患は、球脊髄性筋萎縮症、脊髄性筋萎縮症、ALS8型、運動ニューロン障害VIIB型、原発性側索硬化症、遺伝性痙性対麻痺、ヘキソサミニダーゼA欠乏症、成人ポリグルコサン小体病、BSCL2遺伝子関連神経疾患、骨パジェット病と前側頭葉型痴呆を伴う遺伝性封入体筋炎など多数です。また、ALSと鑑別が必要な後天性疾患も多数あり、頚椎疾患、脳幹や脊髄の腫瘍、甲状腺疾患、鉛中毒、ビタミンB12欠乏症、多発性硬化症、潜在癌による腫瘍随伴症候群、運動ニューロパチー、重症筋無力症、筋無力症候群、封入体筋炎などです。
どのような治療が行われるの?
ALSは、まだ原因が完全に解明されていないため、病気を根本的に直す治療は行われていません。ALSの進行を遅らせる薬として、日本では今、リルゾール(商品名:リルテック)という飲み薬が治療に使われています。この薬は、グルタミン酸の作用をブロックする薬で、グルタミン酸毒性を低減させることが期待されます。リルゾールの欧米における治験では、わずかに生存期間を延長させる効果が示されています。また、エダラボン(商品名:ラジカット)という、フリーラジカルを除く目的の注射薬も、進行を遅らせる薬として治療に使われています。リルゾールもエダラボンも、保険がききます。他にも、病気の進行を遅らせるための薬の開発が複数進んでおり、実際に治験が始まっていたり、これから始める準備が行われていたりする状況です。
その他は、進行に伴って出現するさまざまな症状を軽減するための「対症療法」が行われます。例えば、筋肉や関節の痛みを軽減するためのリハビリテーション、痛みそのものに対する鎮痛薬や湿布薬、体が思うように動かなかったり不安だったりして眠れない場合には睡眠薬や安定薬、呼吸困難に対しては鼻マスクや気管切開、人工呼吸器の使用などです。在宅で人工呼吸器やたんの吸引機などを使用する場合には、自然災害時などに停電した場合の電源確保の方法を、あらかじめ考えておくことが重要です。
また、飲み込みにくい場合(嚥下障害)には、食べ物を柔らかくしたり、とろみをつけたり、少しずつ食べるようにして工夫しますが、進行した場合には、胃ろうや流動食、点滴による栄養補給なども行われます。飲み込みづらかったり、手が動かしにくかったりすると、食べるのが億劫になる人もいますが、栄養や水分の摂取は生きていく上で必須です。ぜひ、あきらめずに、看護師さんなどにコツを聞きながら、栄養不足や水分不足を防ぐように心がけてください。
話しにくくなったり、手の力が入らなくなったりして、家族や周囲の人とのコミュニケーションが難しくなってきた場合には、コミュニケーションボード(文字盤など)や、コンピュータを用いた意思伝達装置などを使用する方法があります。こうした新しいコミュニケーション手段を使用するには、時間をかけた練習が必要なので、症状が進行する前に、早めに主治医の先生や自治体の保健師さんなどに相談してみましょう。
ALSは、長期の療養が必要な病気です。神経内科医、専門看護師、呼吸器科医、言語療法士、理学療法士、作業療法士、呼吸療法士、栄養士、臨床心理士、ソーシャルワーカー、遺伝専門医など、多数の専門家により構成される医療チームのケアを受けていくのが、患者さんにとって望ましいという意見もあります。
今後、新しく治療法を開発していくために、厚生労働省は、ALS患者さんを対象に、JaCALSという全国調査を行っています。JaCALSでは、遺伝子を含む患者さんのさまざまな臨床情報を大規模に調査する研究で、この研究によって、ALSになる仕組みが解明されたり、新しい治療法につながっていったりする可能性があります。この調査には、ALSと診断され告知されている全ての患者さんが参加でき、実際に1,000人以上の人が登録して参加しています。できるだけ大勢の人が参加することが、正しい調査データにつながり、新しい治療法開発へのスピードにもつながってきます。JaCALSへの参加方法については、こちらをご参照ください。
2024年12月27日、「クアルソディ(R)髄注100mg」(一般名:トフェルセン)が、SOD1遺伝子変異を有する筋萎縮性側索硬化症における機能障害の進行抑制を適応症として、国内での製造販売承認を受けました。
どこで検査や治療が受けられるの?
日本で筋委縮性側索硬化症の診療を行っていることを公開している、主な施設は以下です。
- 東北大学病院
- 仙台西多賀病院 筋・神経センター
- 自治医科大学附属病院 脳神経センター内科部門(脳神経内科)
- 東京都立神経病院
- 東京女子医科大学 神経内科
- 国立精神・神経医療研究センター
- 順天堂大学 脳神経内科
- 千葉大学医学部附属病院 脳神経内科
- 東名古屋病院 脳神経内科
- 愛知医科大学病院
- 三重大学医学部附属病院 脳神経内科
- 京都大学医学部附属病院 脳神経内科
- 京都府立医科大学附属病院 脳神経内科
- 関西医科大学附属病院 脳神経内科
- 大阪大学医学部附属病院 神経内科・脳卒中科
- 徳島大学病院 脳神経内科
- 松江医療センター 呼吸器センター
- 九州大学病院 脳神経内科
- 鹿児島大学病院 脳神経内科
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
筋萎縮性側索硬化症の患者会で、ホームページを公開しているところは、以下です。
参考サイト
・参考文献:医学書院 医学大辞典 第2版







