



どのような病気?
遺伝子変異が原因で、発熱などの炎症症状が起こってくる遺伝性疾患です。現時点で対象となるのは、NLRC4異常症、アデノシンデアミナーゼ-2(ADA2)欠損症、エカルディ・グティエール症候群(AGS)、A20ハプロ不全症(HA20)の4疾患です。ただし、次々と新しい自己炎症性疾患が見つかっており、今後対象となる疾患が増えていく可能性があります。
遺伝性自己炎症性疾患に含まれる疾患
疾患名 | 原因遺伝子 | 症状 |
|---|---|---|
NLRC4異常症 | NLRC4 | 発熱、蕁麻疹、関節痛、腸炎(乳児期)、脾臓の腫れ、血球の減少、血液凝固能の障害、など |
ADA2欠損症 | CECR1 | 発熱、紅斑、蔓状の皮斑、麻痺、痺れ、目の異常、胃腸炎、筋肉痛、関節痛、高血圧、腎障害、白血球減少、貧血、血小板減少、自己免疫疾患、など |
エカルディ・グティエール症候群 | TREX1、RNASEH2A、RNASEH2B、RNASEH2C、SAMHD1、ADAR、IFIH1 | 発熱、てんかん、発達退行、肝臓や脾臓の腫れ、肝酵素の上昇、血小板減少、しもやけのような皮膚病変、自己免疫疾患、など |
A20ハプロ不全症 | TNFAIP3 | 発熱、口腔内アフタ、皮疹、関節痛、外陰部潰瘍、消化管潰瘍、ぶどう膜炎、など |
NLRC4異常症
長期間、発熱と平熱を繰り返す、寒冷刺激により誘発される蕁麻疹、関節痛、脾腫(脾臓が腫れる)、血球減少、凝固障害といったマクロファージ活性化症候群など、さまざまな症状が現れます。新生児・乳児期に腸炎(乳児期発症腸炎)を発症することがあります。難聴、無菌性髄膜炎などを示す症例も報告されています。
マクロファージ活性化症候群を合併すると、急激な血球減少、高フェリチン血症、凝固異常によって、生命の危険を伴うことがあります。乳児期発症腸炎は胎児期の発症や重症化するケースが報告されていますが、乳児期を過ぎると徐々に軽快する傾向があります。成人後の予後については、データが少なく詳細は判明していません。
日本では、これまで3家系で報告されています。患者さんの数は十数人と推定されています。
ADA2欠損症
主に血管に炎症が起こり、繰り返す発熱、紅斑や蔓状の皮斑、レイノー症状(寒冷刺激で手足の先が青白く変色する症状)のほか、神経症状(まひや痺れなど)、胃腸炎症状、筋肉痛、関節痛、高血圧、腎障害など多彩な症状が現れます。静脈閉塞や視神経萎縮、脳神経麻痺などによって目に症状が出ることがあります。また、白血球減少や貧血、血小板減少、低ガンマグロブリン血症、自己免疫性リンパ増殖性疾患様症状などが認められます。易感染性(感染しやすい状態)を示すこともあります。
学童期までに発症することが多いとされますが、同じ遺伝子変異を持つ人同士、さらにそれが同じ家系内であっても症状や発症年齢に大きな差が認められることが知られています。ADA2欠損症の症状は長期間にわたって続き、血管炎症による脳梗塞や神経障害、視力障害、臓器梗塞による腎症などの合併は、経過や予後に影響するとされています。長期的な予後については不明な点が多いですが、成人後に発症した症例も報告されていることから、原因遺伝子に変異がある人は若年期に無症状であっても、生涯にわたり通院して診療を受けることが必要になります。
日本では、患者さんは十数人程度と推定されています。
エカルディ・グティエール症候群
重度の心身障害をきたす早期発症型の脳症で、てんかんや発達退行、易刺激性(ささいなことで不機嫌になる)、進行性の小頭症を中心とした神経学的異常が現れます。加えて、肝脾腫(肝臓と脾臓が腫れる)、肝逸脱酵素(肝臓が障害されると血液中に漏れ出すタンパク質)の上昇、血小板減少、間欠的発熱(発熱と平熱を繰り返す)といった先天性感染症(母親の感染が原因で赤ちゃんが感染した状態で生まれること)に似た症状も見られます。また、約4割の人で手足の指や耳などに凍瘡(しもやけ)のような皮膚病変を伴います。全身性エリテマトーデスに類似した自己免疫疾患の合併を認める場合もあります。
出生時から症状が現れる場合もありますが、発症時期はさまざまです。しばらく正常に発達した後、症状が目立ってくることもよくあります。海外の報告では、約20%の人は出生前に発症が始まり、成長の遅れ(子宮内発育遅延)と脳の異常、特に脳の石灰化が超音波画像で確認される場合があるとされます。従来は生後数年以内に命を落とすと考えられていましたが、近年では軽症例の報告も増えています。
国内の患者さんの数は、数十人程度と推定されています。
A20ハプロ不全症
周期的あるいは持続的な発熱や、口腔内アフタ(口内炎に似た潰瘍)、皮疹、関節痛に加え、外陰部潰瘍、消化管潰瘍、ぶどう膜炎といったベーチェット病に似た症状が見られます。一部の人では、自己免疫疾患(自己免疫性甲状腺炎、Ⅰ型糖尿病、自己免疫性肝炎、ループス腎炎、IgA血管炎など)、ネフローゼ症候群の合併が報告されています。
新生児期から29歳までの比較的若年期に発症することが知られています。生涯にわたって全身の炎症が続くため、生活の質(QOL)低下が課題となります。長期的な予後に関しては、まだ不明な部分が多いですが、ベーチェット病に似た症状が年齢とともに徐々に現れてくることがあるので、眼科診察、検便(便潜血)などは定期的に受ける方がよいとされます。また、A20ハプロ不全症では、自己免疫疾患発症のリスクが指摘されており、甲状腺機能の異常、血糖値の異常などにも注意する必要があります。
A20ハプロ不全症の患者さんは、日本に数十人程度存在すると推定されています。
これら4疾患を含む遺伝性自己炎症疾患は、指定難病対象疾病(指定難病325)および、小児慢性特定疾病の対象疾患となっています。
何の遺伝子が原因となるの?
遺伝性自己炎症性疾患の原因となる遺伝子は、それぞれの疾患によって異なります。
NLRC4異常症
NLRC4異常症の原因遺伝子として、「NLRC4遺伝子」が見つかっています。NLRC4遺伝子は「NLRC4タンパク質」の設計図となる遺伝子です。NLRC4タンパク質は炎症反応を制御する役割を持つ「インフラマソーム」と呼ばれる構造体を作るタンパク質のひとつです。NLRC4異常症の患者さんでは、NLRC4遺伝子の変異によって、異常に活性化されたNLRC4タンパク質が作られるようになります。その結果、炎症を誘発する生体物質であるインターロイキン(IL-1β、IL-18)が過剰産生され、炎症が起こります。
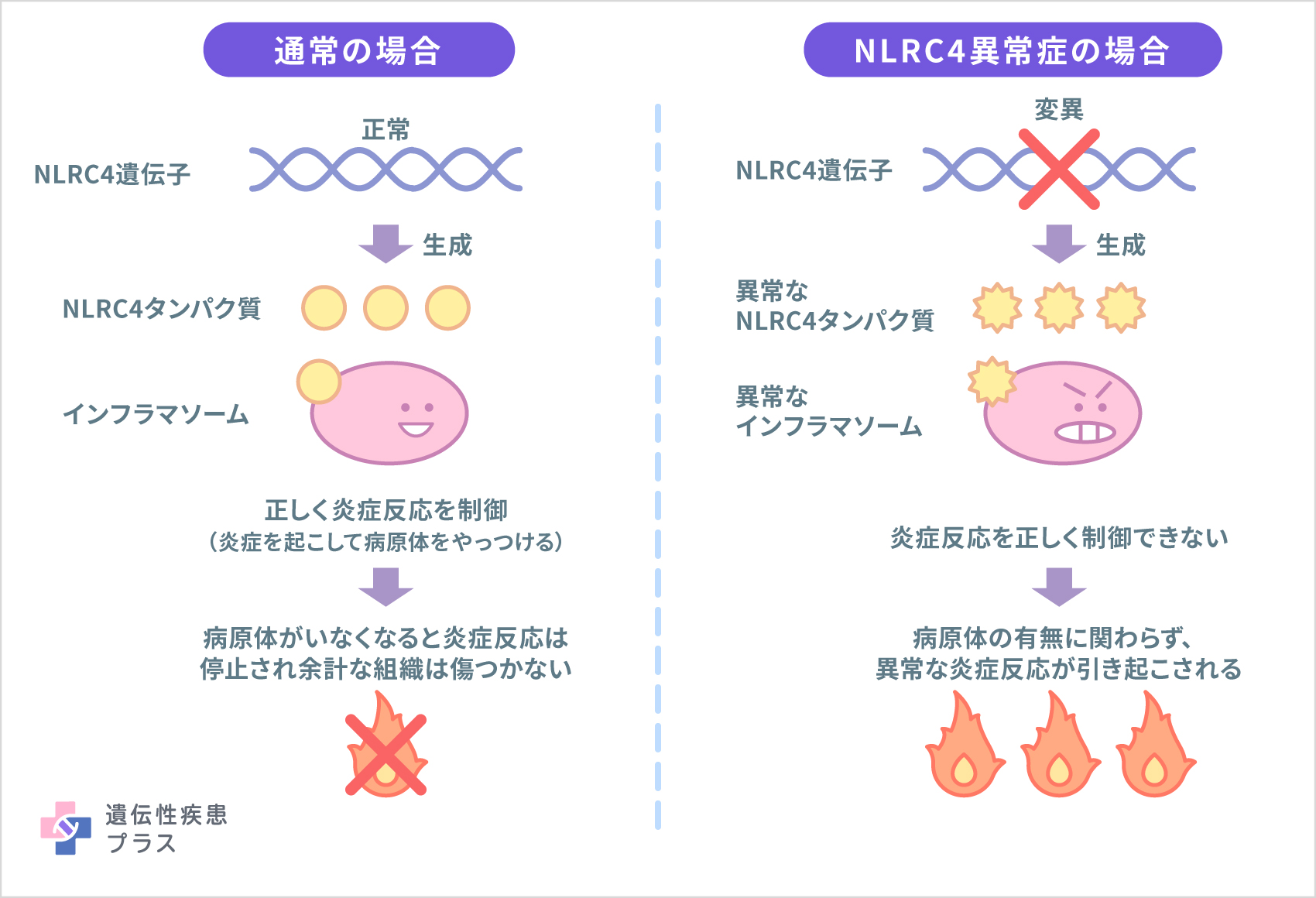
NLRC4異常症は、常染色体優性(顕性)遺伝と呼ばれる形式で遺伝します。常染色体優性(顕性)遺伝形式では、両親のどちらかがこの病気の場合、子どもは50%の確率で発症します。ただし、多くの場合は、本人以外に家族の中で発症した人がいない孤発例として報告されています。また、体細胞モザイク(変異のある体細胞と変異のない体細胞が混ざった状態)の例も見られます。

ADA2欠損症
ADA2欠損症は、「CECR1遺伝子」の変異によって発症します。CECR1遺伝子から「ADA2タンパク質」が作られます。ADA2タンパク質は体内でアデノシンという物質をイノシンという物質に分解する酵素で、ADA2欠損症の患者さんでは血液中に存在するADA2タンパク質の酵素活性が低下または消失します。
ADA2欠損症の発症メカニズムは完全には解明されていませんが、ADA2タンパク質の活性低下/消失によるアデノシン濃度の慢性的な上昇が血管の炎症を促進する可能性が指摘されています。
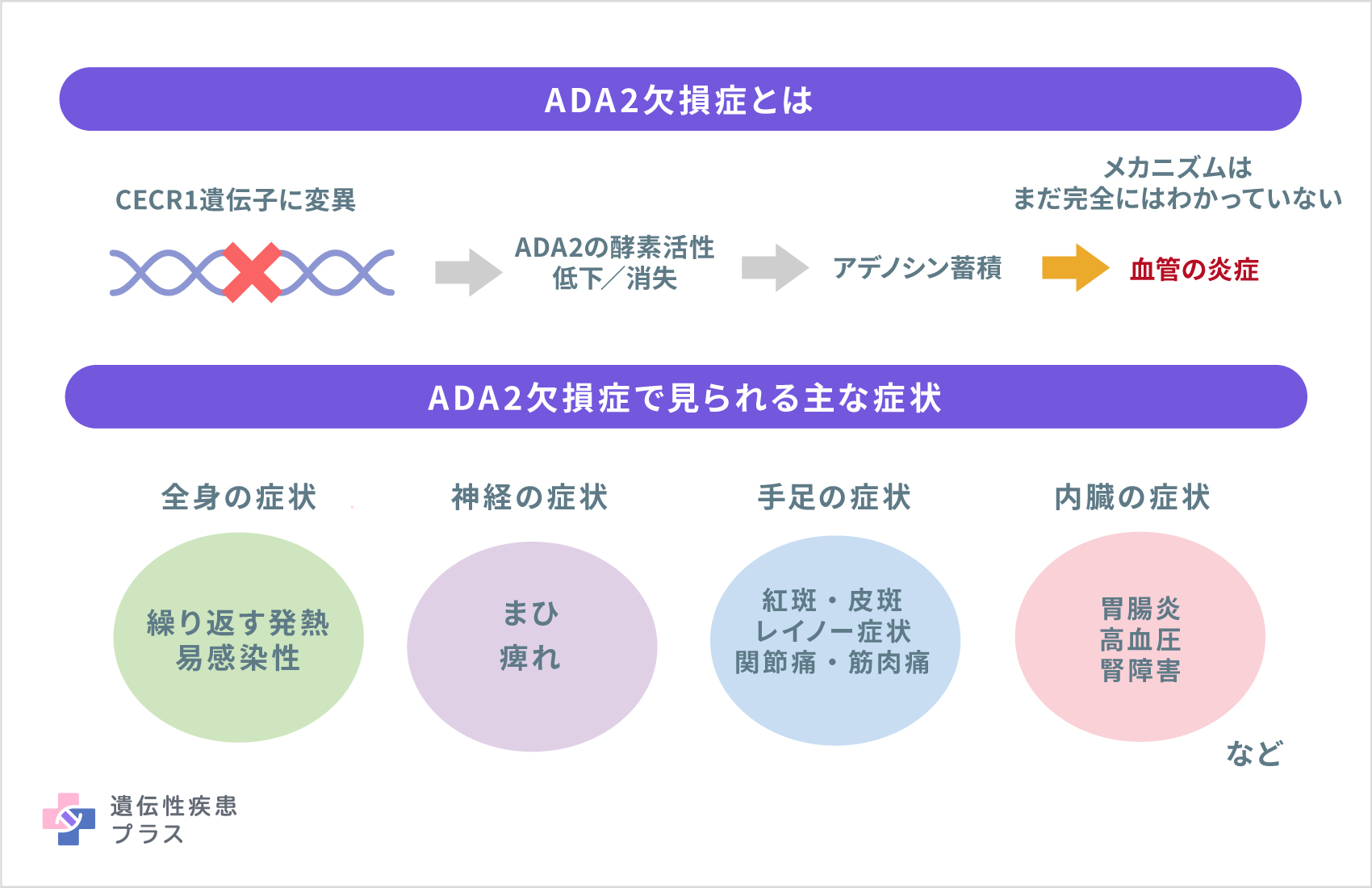
CECR1遺伝子の変異は、常染色体劣性(潜性)遺伝と呼ばれる遺伝形式で遺伝します。この場合、それぞれの細胞が2つずつ持つ遺伝子の両方に変異が生じることで発症します。両親は変異のある遺伝子を1つ持つ保因者ではありますが、通常はこの病気を発症しません。保因者である両親から生まれた子どもが発症する確率は25%です。

エカルディ・グティエール症候群
原因遺伝子として、今のところ「TREX1」「RNASEH2A」「RNASEH2B」「RNASEH2C」「SAMHD1」「ADAR」「IFIH1」の7つが報告されています。これらの遺伝子はいずれも核酸(DNAとRNA)の代謝や細胞内の核酸を検知する仕組みに関わっており、遺伝子変異によってI型インターフェロン(IFN)の過剰産生と異常な免疫反応が引き起こされます。また、米国国立医学図書館が運営するMedline Plusによると、RNU7-1遺伝子やLSM11遺伝子など、他の遺伝子の変異も病気を引き起こす可能性があるとされます。
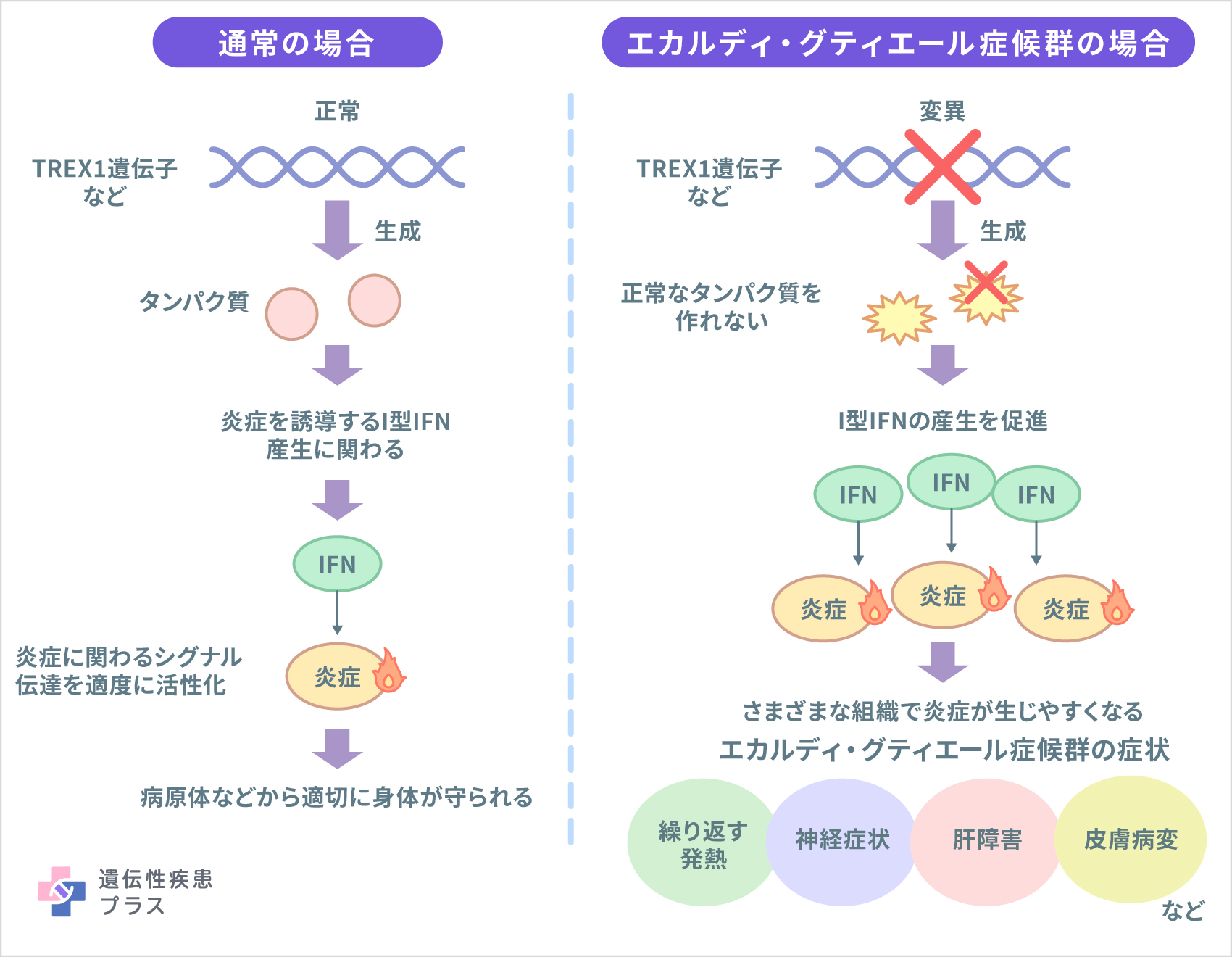
遺伝形式は遺伝子によって異なります。TREX1遺伝子、RNASEH2A遺伝子、RNASEH2B遺伝子、RNASEH2C遺伝子、SAMHD1遺伝子、ADAR遺伝子およびRNU7-1遺伝子の変異は常染色体劣性(潜性)遺伝形式で遺伝します。一方、IFIH1遺伝子の変異、およびTREX1遺伝子、ADAR遺伝子の一部の変異は、常染色体優性(顕性)遺伝形式で遺伝することが報告されています。
A20ハプロ不全症
原因として「TNFAIP3遺伝子」の変異が知られています。TNFAIP3遺伝子は「A20」というタンパク質の設計図です。A20には炎症を調節する働きがあることが知られています。患者さんの細胞は、2つ持つTNFAIP3遺伝子のうち片方に変異があるため、正常に機能するA20を十分な量作ることができません(ハプロ不全)。そのため、過剰な炎症反応が全身に起こります。
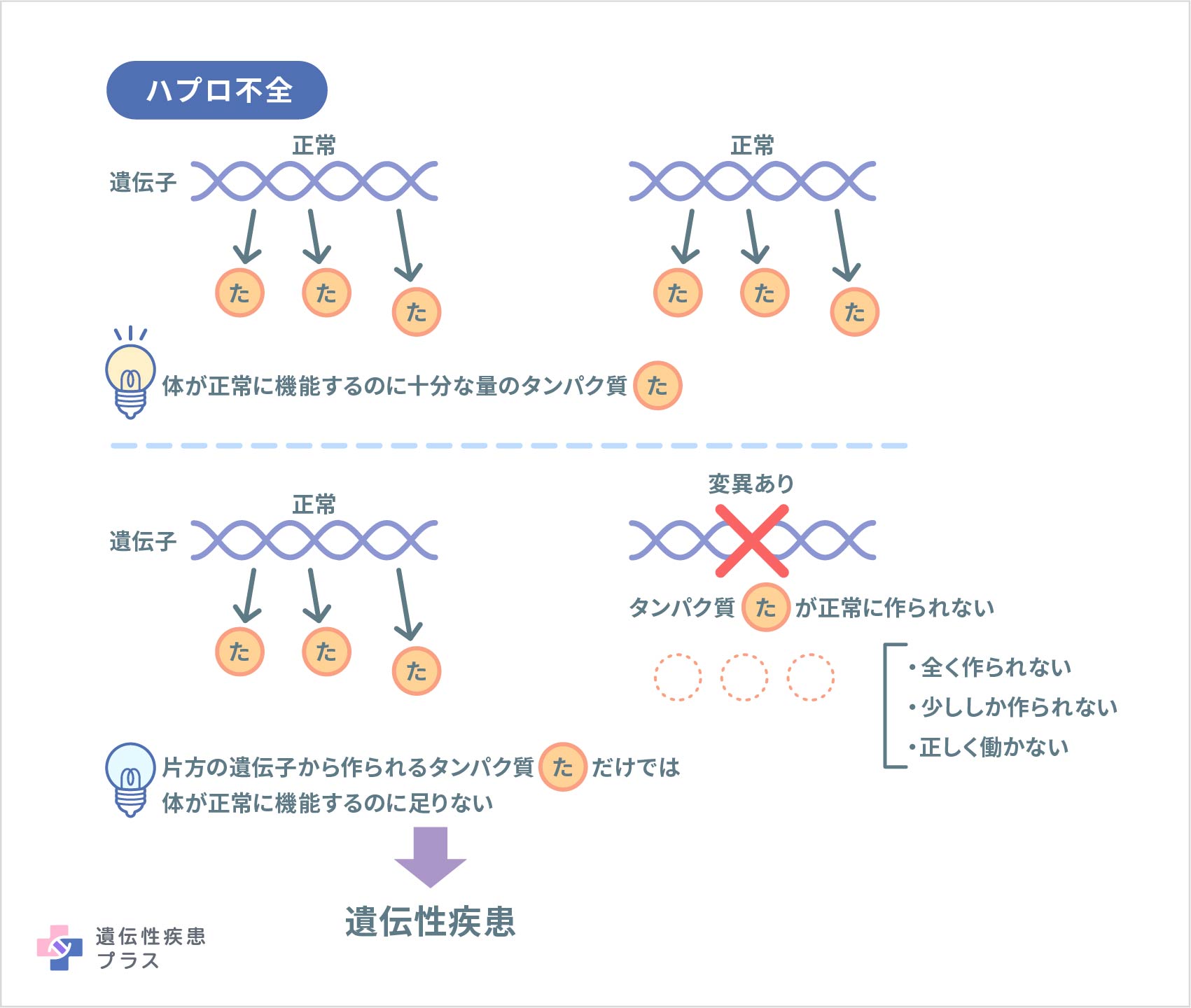
この病気は、常染色体優性(顕性)遺伝形式で発症するとされます。
どのように診断されるの?
難病情報センターのウェブサイトで、遺伝性自己炎症疾患に含まれる4つの対象疾患それぞれについて診断基準が公開されています。症状、検査所見、遺伝学的検査において該当する項目によって、Definite(確定)、Probable(可能性が高い)と判定され、さらに鑑別すべき疾患に該当しないことが確認された場合に遺伝性自己炎症疾患と診断されます。
NLRC4異常症
NLRC4異常症では、白血球数の上昇、CRP高値、赤血球沈降速度(赤沈)の上昇などが見られます(いずれも炎症を示す所見)。マクロファージ活性化症候群を合併した人では、血小板減少、白血球減少、貧血、高フェリチン血症、凝固異常が認められます。血清中の総IL-18濃度は104 pg/mL以上の高い値を示すことが多いとされます。
<診断基準>
A.症状
①紅斑、蕁麻疹様発疹
②発熱
③持続する下痢等の腸炎症状
B.検査所見
①炎症所見陽性
②血清IL-18高値
③マクロファージ活性化症候群
C.遺伝学的検査
NLRC4遺伝子に疾患関連変異を認める。
<診断のカテゴリー>
Definite:Aの2項目+Bの2項目+Cを満たすもの
Probable:
(1)Aの2項目+Bの1項目+Cを満たすもの
(2)Aの1項目+Bの2項目+Cを満たすもの
(3)Aの1項目+Bの1項目+Cを満たすもの
<鑑別すべき疾患>
他の自己炎症性疾患、全身型若年性特発性関節炎、慢性感染症、炎症性腸疾患、リウマチ・膠原病疾患、家族性血球貪食性リンパ組織球症、X連鎖性リンパ増殖症
ADA2欠損症
画像検査で、虚血性梗塞や出血性梗塞、動脈瘤の存在が確認できます。組織検査では、好中球性の炎症所見や血管炎が認められます。血液検査では、白血球減少(好中球、リンパ球、単球)、貧血、血小板減少、低ガンマグロブリン血症、あるいは炎症反応の上昇が見られます。
<診断基準>
A.症状
①繰り返す発熱
②蔓状皮斑やレイノー症状などの皮膚症状
③麻痺(まひ)や痺れなどの神経症状
B.検査所見
①画像検査:虚血性(時に出血性)梗塞や動脈瘤の存在
②組織検査:血管炎の存在
③ADA2活性検査:血漿中ADA2酵素活性の明らかな低下
C.遺伝学的検査
CECR1遺伝子に機能喪失型変異をホモ接合又は複合型ヘテロ接合で認める。
<診断のカテゴリー>
Definite:Aの1項目+B①またはB②+B③またはCのいずれかを満たすもの
Probable:Aの1項目+B③またはCのいずれかを満たすもの
<鑑別すべき疾患>
他の自己炎症性疾患、全身型若年性特発性関節炎、慢性感染症及びベーチェット病・高安動脈炎などの非遺伝性血管炎症候群
エカルディ・グティエール症候群
検査における特徴は、頭蓋内石灰化病変と慢性的な髄液細胞数、髄液インターフェロン-α、髄液ネオプテリンの増加です。ただし、髄液に異常が見られるのは、生後数年以内のみであることが多いとされます。血小板減少、肝脾腫、肝逸脱酵素の上昇、間欠的発熱などから不明熱として精密検査が行われることがよくありますが、感染に関わる指標には基本的に異常がありません。IgM、IgGの上昇と補体の低下も報告されています。
<診断基準>
A.症状
①神経症状(早発性脳症、発達遅滞、進行性の小頭症、痙攣)
②神経外症状(不明熱、肝脾腫、凍瘡様皮疹)
B.検査所見
①髄液検査異常(ア~ウの1項目以上)
ア)髄液細胞数増多(WBC≧5/mm3、通常はリンパ球優位)
イ)髄液中インターフェロンα上昇(>6IU/mL)
ウ)髄液中ネオプテリン増加(年齢によりカットオフ値は異なる)
②画像検査所見:頭蓋内石灰化(加齢による生理的変化を除く)
C.遺伝学的検査
TREX1、RNASEH2B、RNASEH2C、RNASEH2A、SAMHD1、ADAR、IFIH1等の疾患原因遺伝子のいずれかに疾患関連変異を認める。
<診断のカテゴリー>
Definite:Aの①+B①およびB②+Cのいずれかを満たすもの
Probable:
(1)Aの1項目+B②+Cのいずれかを満たすもの
(2)Aの①+B①およびB②を満たすもの
<鑑別すべき疾患>
他の自己炎症性疾患、全身型若年性特発性関節炎、慢性感染症、リウマチ・膠原病疾患、サイトメガロウィルス・風疹・トキソプラズマ・単純ヘルペス・HIVを含む出生前/周産期感染症、既知の先天代謝性疾患・脳内石灰化症・神経変性疾患
A20ハプロ不全症
検査所見として、症状が全身に現れている場合、白血球数、CRP、血清アミロイドA、赤沈などの炎症反応の上昇が見られます。症状が口内炎のみなど軽微な場合、血液検査による炎症反応の上昇を確認できないこともあります。消化管でのベーチェット病様症状の有無を確認するには、便潜血検査、消化管内視鏡検査、PET-CT検査などが有用です。また、ベーチェット病同様に髄液中のIL-6濃度が高くなったり、針反応試験が陽性になったりすることがあります。なお、遺伝子変異が同定されても臨床症状を伴わない場合は、A20ハプロ不全症とは診断されません。
<診断基準>
A.症状
①反復性発熱
②反復性口腔内アフタ
③下痢、血便等の消化管症状
④外陰部潰瘍
⑤関節炎
⑥皮疹(毛嚢炎様皮疹、痤瘡様皮疹、結節性紅斑様皮疹など)
⑦眼症状(虹彩毛様体炎、網膜ぶどう膜炎など)
⑧自己免疫疾患症状(自己免疫性甲状腺炎、自己免疫性肝炎など)
B.検査所見
①炎症所見陽性
②便潜血陽性
③針反応試験陽性
C.遺伝学的検査
TNFAIP3遺伝子に疾患関連変異を認める。
<診断のカテゴリー>
Definite:Aの2項目+Bの1項目+Cを満たすもの
Probable:Aの1項目+Cを満たすもの
<鑑別すべき疾患>
他の自己炎症性疾患(家族性地中海熱、クリオピリン関連周期熱症候群、TNF受容体関連周期性症候群、中條-西村症候群、PAPA症候群、Blau症候群/若年発症サルコイドーシス、高IgD症候群/メバロン酸キナーゼ欠損症、PFAPA症候群)、若年性特発性関節炎、慢性感染症、炎症性腸疾患、悪性新生物、リウマチ・膠原病疾患、ベーチェット病
どのような治療が行われるの?
いずれの疾患も標準的な治療はまだ確立されてはいませんが、それぞれの疾患で有効性が報告されている治療法がいくつかあります。これらの治療法の中には、未承認、適用外薬も含まれるため、治療にあたっては専門の医療機関に相談することが重要です。
NLRC4異常症
軽症例では、非ステロイド性抗炎症薬(NSAIDs)による治療が検討されます。重症例では、抗体医薬である抗IL-1製剤(カナキブマブ)の使用が考慮されることがあります。
症例報告レベルでは、寒冷蕁麻疹(かんれいじんましん)などの皮膚症状に対してはNSAIDs、国内未承認薬であるアナキンラ(IL-1受容体拮抗薬)が有効であったとされます。乳児期発症腸炎を合併しない患者さんでは、抗IL-1製剤も皮膚症状に対して有効であると考えられています。
乳児期発症腸炎については、腸内細菌叢の関与が推定されていますが、現時点では糞便移植による治療は推奨されていません。
マクロファージ活性化症候群に対しては、ステロイド、シクロスポリン、免疫グロブリン療法の併用が有効との報告があります。研究段階ではありますが、マクロファージ活性化症候群の症状に対し、IL-18結合タンパク質の有効性が示唆されています。
NLRC4タンパク質は骨髄細胞や腸管上皮細胞に発現するため、造血細胞移植あるいは腸管移植の治療効果は期待されていません。
寒冷刺激は炎症を誘発する可能性があることから、冬季の保温とともに、夏のエアコンなどによる急激な温度低下もできるだけ避けることが推奨されます。
ADA2欠損症
ほとんどの患者さんでは、副腎皮質ステロイド(中等量~高容量)によって症状が改善しますが、薬を減らすと再び悪化する例が多く見られます。免疫抑制剤や免疫グロブリン補充療法が併用されますが、効果が見られないことが、しばしばあります。
現時点で全身炎症や血管の炎症症状に対して最も効果が期待されるのは抗TNF製剤(インフリキシマブ、アダリムマブ、エタネルセプト)です。ただし、血液学的症状(貧血、白血球減少、血小板減少など)への有効性は定まっていません。また、抗TNF製剤そのものは承認済みですが、ADA2欠損症に対しては、現時点では保険適用されていません。
ADA2欠損症に対して造血幹細胞移植を行った例が報告されており、今のところ唯一の根治療法と考えられています。血漿交換や血漿輸血などによる血中ADA2の補充に関しては、有効性が定まっていません。また、血漿交換や血漿輸血以外のADA2補充療法は、今のところ存在しません。
無症状例および臨床症状が軽微な症例に対する治療方針について、現時点で定まったものはありません。しかしながら、発熱などの明らかな全身炎症を伴わなくとも、脳梗塞などの症状が突然現れる可能性があるため、慎重な経過観察が望ましいとされます。
エカルディ・グティエール症候群
治療は対症療法が中心で、神経症状はリハビリテーションや医療的ケアの対象となります。炎症症状や合併する自己免疫性疾患に対しては、免疫抑制療法や免疫修飾療法が有効な可能性があります。寒冷刺激が炎症を誘発することがあるため、冬だけでなく、夏もエアコンなどによる急激な温度低下に注意が必要です。
凍瘡様皮疹に対しては、四肢末端の保温、ステロイド軟膏、カルシウム拮抗剤などが使用されます。治療法として確立されてはいませんが、JAK阻害薬であるバリシチニブによる症状と検査値の改善、逆転写酵素阻害薬(ウイルスの増殖を抑える薬)による血液・髄液検査所見の改善が報告されています。
A20ハプロ不全症
炎症症状に対しては、NSAIDs、副腎皮質ステロイド全身投与、コルヒチン、抗TNF製剤(エタネルセプト、インフリキシマブ、アダリムマブなど)などの使用が報告されています。ただし、これらの治療法の有効性は確立していません。
治療が難航した腸管炎症に対して、腸管切除が行われることがあります。また、難治性自己免疫疾患多発症例では、骨髄移植が有効であった一例が報告されています。ネフローゼ症候群を合併した患者さんに対して、リツキシマブが使用されることがあります。その他、自己免疫疾患を合併した場合には、それぞれの疾患に合わせた治療を行う必要があります。
どこで検査や治療が受けられるの?
日本で遺伝性自己炎症疾患の診療を行っていることを公開している、主な施設は以下です。
- 信州大学医学部附属病院 リウマチ・膠原病内科
- 東京科学大学病院 遺伝子診療科
- 横浜市立大学附属病院 血液・リウマチ・感染症内科
- 岐阜大学医学部附属病院 小児科
- 京都大学医学部附属病院 小児科
- 大阪母子医療センター 遺伝診療科
- 大阪医科薬科大学病院 難病総合センター 小児科
- 久留米大学病院 小児科
※このほか、診療している医療機関がございましたら、お問合せフォームからご連絡頂けますと幸いです。
患者会について
難病の患者さん・ご家族、支えるさまざまな立場の方々とのネットワークづくりを行っている団体は、以下です。
参考サイト
- 難病情報センター
- 小児慢性特定疾病情報センター 自己炎症性疾患
- 小児慢性特定疾病情報センター エカルディ・グティエール(Aicardi-Goutieres)症候群
- KEGG DISEASE データベース 遺伝性自己炎症疾患
- MedlinePlus
- Genetic and Rare Diseases Information Center
- 厚生労働科学研究費補助金 難治性疾患政策研究事業 自己炎症性疾患とその類縁疾患の全国診療体制整備、重症度分類、診療ガイドライン確立に関する研究 平成29年度~令和元年度 総合研究報告書






